Case Report: Acute Intermittent Porphyria






A 34-year-old pregnant woman presented for evaluation of severe, persistent abdominal pain.
Case
A 34-year-old woman presented to the ED with severe, persistent abdominal pain that had begun 18 days earlier. She was 7 weeks pregnant and had been seen in the same ED the day before. During that visit, ultrasound had shown a single pregnancy of doubtful viability. Abdominal magnetic resonance imaging was normal. She was given multiple doses of hydromorphone. The discharge diagnosis was “missed abortion.”
Since the onset of her pain, she had been hospitalized twice elsewhere, with no clear diagnosis to explain her pain. Treatment consisted of repeat doses of hydromorphone. During the second hospitalization, a sodium level of 109 mEq/L had been corrected with hypertonic saline, and a urinary tract infection (UTI) had been treated with cephalexin.
Our patient had never experienced similar abdominal pain. Her medical history included depression and asthma. Her family history was notable for an aunt who had died of lung cancer.
On this ED visit, the patient’s vital signs were normal. On examination, she was moaning in pain and clutching her abdomen. The abdomen was tender in both lower quadrants, with guarding but no rebound. Her sodium level was 125 mEq/L; the day before it had been 132 mEq/L.
Urine dipstick testing showed 2+ glucose and 2+ bilirubin; both had been within normal range (negative) the day before. An abdominal/pelvic computed tomography scan with intravenous (IV) and oral contrast did not reveal any potential cause of the patient’s pain. Multiple doses of IV hydromorphone were given, but her pain persisted.
We revisited our patient’s family history. With prompting, she remembered that as a teenager, her mother had had an illness that caused “problems with her nerves and blood vessels and turned her urine red.” When reached by phone, the patient’s mother, who lives outside the United States, said she was familiar with the term “porphyria,” but curiously, she did not state she carried the diagnosis, and had not advised her children they could be at risk.
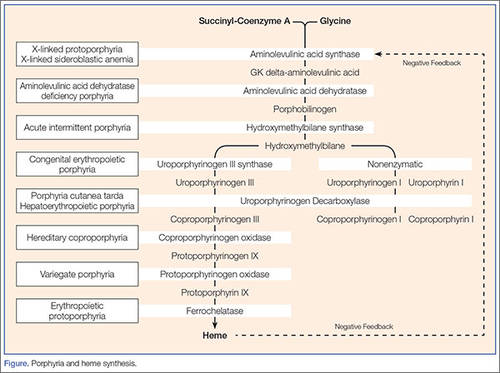
The patient was admitted to the intensive care unit (ICU) for treatment of hyponatremia. Her mother’s history led us to suspect porphyria, so we sent a urine sample from the ED for porphobilinogen (PBG) testing. Her urine was not red at that time (on further questioning, she remembered she had had an episode of “red urine” recently). Two days later, after the PBG result came back positive, treatment was initiated for porphyria. With further stool and serum testing, the diagnosis of acute intermittent porphyria (AIP) was made.
The patient was treated with glucose loading and hemin therapy. In the ICU, 2 ampules of 50% dextrose in water solution (D50W) was administered, and she was transferred to the hematology-oncology service for hemin therapy. Soon after, she underwent a dilation and curettage. Three weeks later, she was discharged in good condition.
Discussion
The unifying diagnostic concept is that toxins damage all components of the nervous system: intestinal, central, peripheral, and autonomic. Hence, any combination of abdominal pain, vomiting, psychiatric symptoms, vital sign instability, weakness, or sensory loss may occur.1 To further confuse the diagnostician, the constellation of symptoms may vary with each episode.
Two critical laboratory clues are red urine—which often is mistaken for a UTI or hematuria—and hyponatremia.1 Another porphyria hallmark is triggers. These include drugs, carbohydrate deprivation, smoking, and stress. Common chemical inciters include alcohol, ketamine, etomidate, macrodantin, nifedipine, progesterone, and phenytoin.1 The Atkins diet (zero carbohydrates) reportedly caused an uptick in new porphyria cases.1
Attacks usually start after puberty. Women tend to experience flares during the luteal (progesterone) phase of the menstrual cycle.1 Acute intermittent porphyria can mimic Guillain-Barré syndrome and psychosis.2 Delayed diagnosis may lead to irreversible neurological damage or death.2
Despite AIP’s complexity, initial diagnostic testing is simple: a urinary PBG level obtained during an attack is virtually 100% sensitive and specific for AIP and two other acute porphyrias: hereditary coproporphyria (HCP), and variegate porphyria (VP). A positive urine PBG mandates immediate treatment—even while awaiting porphyrin and GK delta-aminolevulinic acid (ALA) levels in stool and serum to identify which porphyria (AIP, HCP, or VP) is present. The fourth (and least common) acute porphyria, ALA dehydratase porphyria (ADP), may produce no PBG elevation and requires separate porphyrin and ALA testing to make the diagnosis.