Spontaneous Repigmentation of Silvery Hair in an Infant With Congenital Hydrops Fetalis and Hypoproteinemia





Silvery hair is a characteristic finding of 3 rare autosomal recessive disorders: Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS). We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have one of these classic causative disorders. The patient was delivered at 35 weeks’ gestation with congenital hydrops fetalis associated with a chromosomal abnormality (46,XY,add[2],[p23]), hypothyroidism, hypoproteinemia, and hypogammaglobulinemia. Over the course of follow-up, spontaneous brown repigmentation of the silvery hair was noted. We concluded that the silvery hair was induced by hypoproteinemia secondary to congenital hydrops fetalis.
Practice Points
- Silvery hair is characteristic of 3 rare autosomal-recessive disorders: Chédiak-Higashi syndrome, Elejalde syndrome, and Griscelli syndrome.
- Hypopigmentation is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes.
- Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
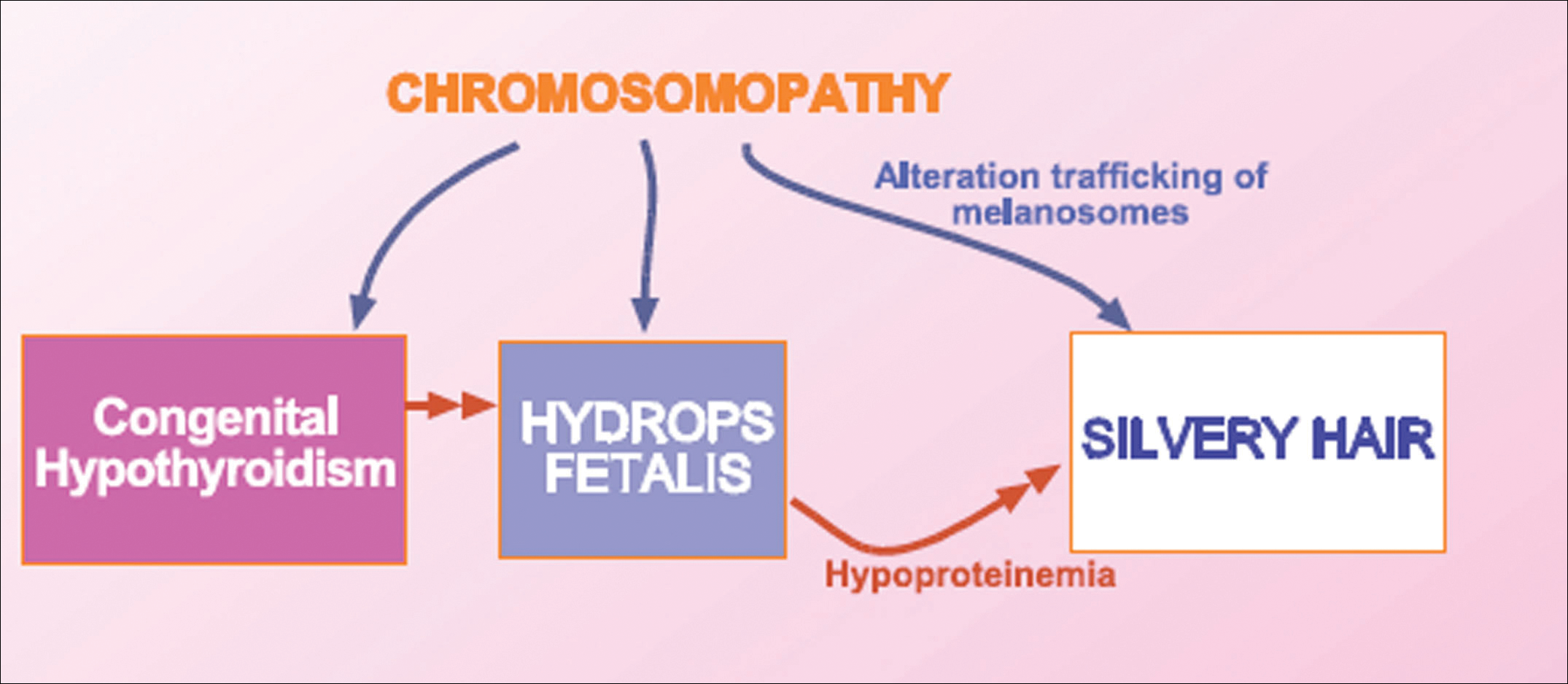
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.