Myasthenia gravis: Newer therapies offer sustained improvement





ABSTRACTMyasthenia gravis is a prototypical antibody-mediated autoimmune neuromuscular disorder. Treatments have improved over the past 30 years, leading to significantly fewer deaths and better quality of life. Future research should further elucidate its pathogenesis, reveal better ways to diagnose it, and yield new treatments.
KEY POINTS
- In most cases of myasthenia gravis, the patient has antibodies against acetylcholine receptor (AChR) or musclespecific tyrosine kinase (MuSK).
- Myasthenia gravis is diagnosed by clinical signs, bedside tests (the ice-pack test or the edrophonium test), serologic tests for AChR antibodies or MuSK antibodies, and electrophysiologic tests.
- Acetylcholinesterase inhibitors are the first-step therapy, but patients who have moderate to severe symptoms require some form of immunomodulating therapy.
- A number of drugs can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. These include penicillamine, interferons, procainamide, quinidine, and antibiotics such as quinolones and aminoglycosides.
Seronegative myasthenia gravis
In a series of 562 consecutive patients with generalized weakness due to myasthenia gravis, 92% were positive for AChR antibody, 3% were positive for MuSK antibody, and 5% were seronegative (possessing neither antibody).15 In contrast, about 50% of patients with purely ocular myasthenia gravis (ie, with isolated weakness of the levator palpebrae superioris, orbicularis oculi, or oculomotor muscles) are seropositive for AChR antibody. Only a few ocular MuSK antibody-positive cases have been described, leaving the rest seronegative. Rarely, both antibodies can be detected in the same patient.16
In patients who are negative for AChR antibodies at the time of disease onset, sero-conversion may occur later during the course. Repeating serologic testing 6 to 12 months later may detect AChR antibodies in approximately 15% of patients who were initially seronegative.15,17
The clinical presentation, electrophysiologic findings, thymic pathologic findings, and treatment responses are similar in AChR antibody-positive and seronegative myasthenia gravis.17 Muscle biopsy study in seronegative cases demonstrates a loss of AChR as well.18
Based on these observations, it has been proposed that seronegative patients may have low-affinity antibodies that can bind to tightly clustered AChRs on the postsynaptic membrane but escape detection by routine radioimmunoassays in a solution phase. With a sensitive cell-based immunofluorescence assay, low-affinity antibodies to clustered AChRs were detected in 66% of patients with generalized myasthenia gravis and in 50% of those with ocular myasthenia gravis who were seronegative on standard assays.19,20 These low-affinity AChR antibodies can also activate complement in vitro, increasing the likelihood that they are pathogenic. However, assays to detect low-affinity AChR antibodies are not yet commercially available.
Within the past year, three research groups independently reported detecting antibodies to LRP4 in 2% to 50% of seronegative myasthenia gravis patients. This wide variation in the prevalence of LRP4 antibodies could be related to patient ethnicity and methods of detection.21–23 LRP4 is a receptor for agrin and is required for agrin-induced MuSK activation and AChR clustering. LRP antibodies can activate complement; therefore, it is plausible that LRP4 antibody binding leads to AChR loss on the postsynaptic membrane. However, additional study is needed to determine if LRP4 antibodies are truly pathogenic in myasthenia gravis.
A DISORDER OF FATIGABLE WEAKNESS

Myasthenia gravis is a disorder of fatigable weakness producing fluctuating symptoms. Symptoms related to the involvement of specific muscle groups are listed in Table 1. Muscle weakness is often worse later in the day or after exercise.
Ocular myasthenia gravis accounts for about 15% of all cases. Of patients initially presenting with ocular symptoms only, twothirds will ultimately develop generalized symptoms, most within the first 2 years.24 No factor has been identified that predicts conversion from an ocular to a generalized form.
Several clinical phenotypes of MuSK antibody-positive myasthenia gravis have been described. An oculobulbar form presents with diplopia, ptosis, dysarthria, and profound atrophy of the muscles of the tongue and face. A restricted myopathic form presents with prominent neck, shoulder, and respiratory weakness without ocular involvement. A third form is a combination of ocular and proximal limb weakness, indistinguishable from AChR antibody-positive disease.25
MuSK antibody-positive patients do not respond as well to acetylcholinesterase inhibitors as AChR antibody-positive patients do. In one study, nearly 70% of MuSK antibody-positive patients demonstrated no response, poor tolerance, or cholinergic hypersensitivity to these agents.25 Fortunately, most MuSK antibody-positive patients have a favorable response to immunosuppressive therapy—sometimes a dramatic improvement after plasmapheresis.8
DIAGNOSIS OF MYASTHENIA GRAVIS
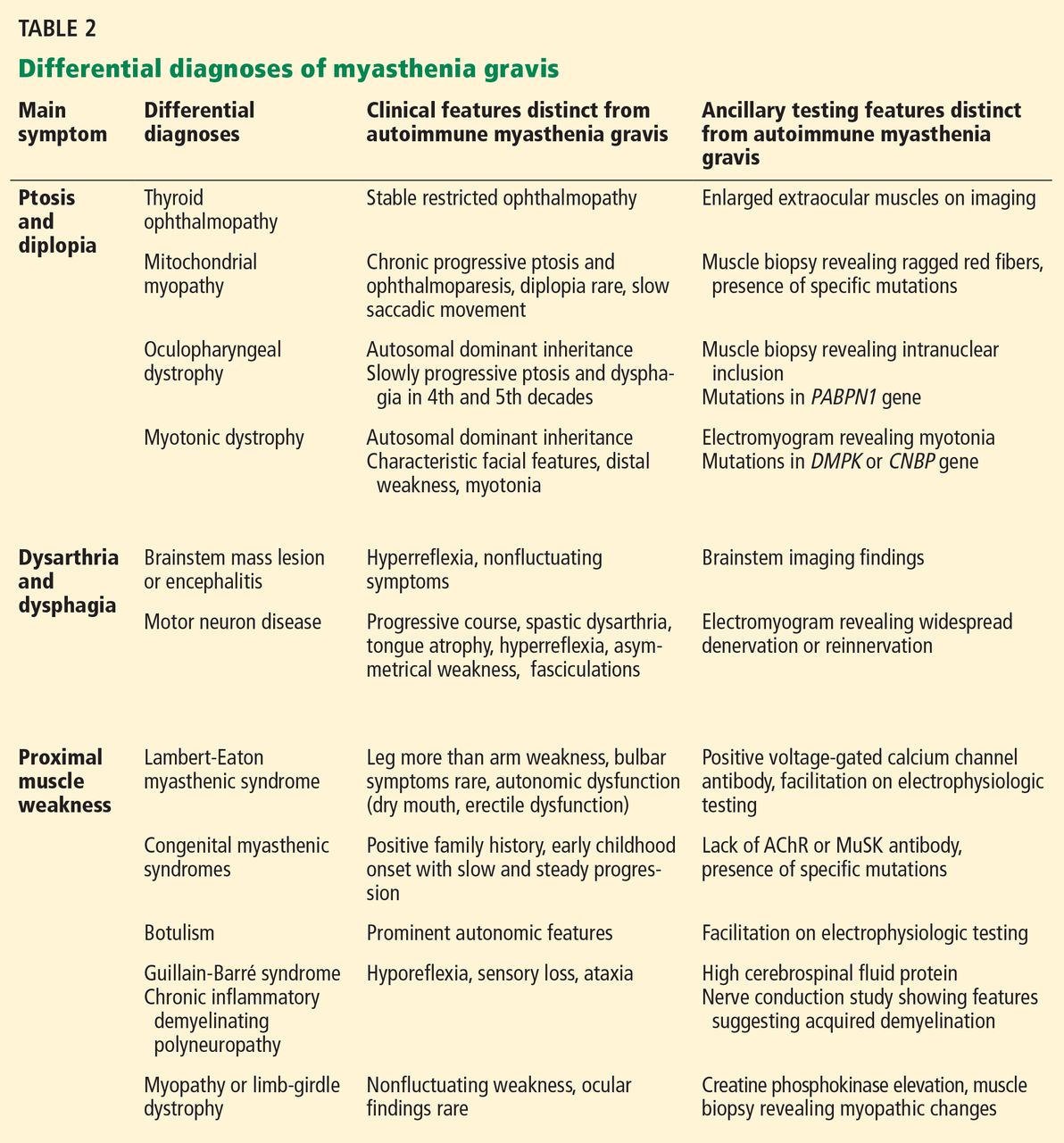
The common differential diagnoses for myasthenia gravis are listed in Table 2.
The essential feature of myasthenia gravis is fluctuating muscle weakness, often with fatigue. Many patients complain of weakness of specific muscle groups after their repeated usage. Pain is generally a less conspicuous symptom, and generalized fatigue without objective weakness is inconsistent with myasthenia gravis.
Signs of muscle weakness may include droopy eyelids, diplopia, inability to hold the head straight, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and difficulty raising the arms or rising from the sitting position. A historical pattern of ptosis alternating from one eye to the other is fairly characteristic of myasthenia gravis.
The weakness of orbicularis oculi is easily identified on examination by prying open the eyes during forced eye closure. Limb weakness is usually more significant in the arms than in the legs. An often-neglected feature of myasthenia gravis is finger extensor weakness with a relative sparing of other distal hand muscles.2
The ice-pack test is performed by placing a small bag of ice over the ptotic eye for 2 to 5 minutes and assessing the degree of ptosis for any noticeable improvement. This test is not very helpful for assessing ocular motor weakness.
The edrophonium (Tensilon) test can be used for patients with ptosis or ophthalmoparesis. Edrophonium, a short-acting acetylcholinesterase inhibitor, is given intravenously while the patient is observed for objective improvement. The patient’s cardiovascular status should be monitored for arrhythmias and hypotension. Atropine should be immediately available in case severe bradycardia develops.
The ice-pack test and the edrophonium test can give false-negative and false-positive results, and the diagnosis of myasthenia gravis must be verified by other diagnostic tests.