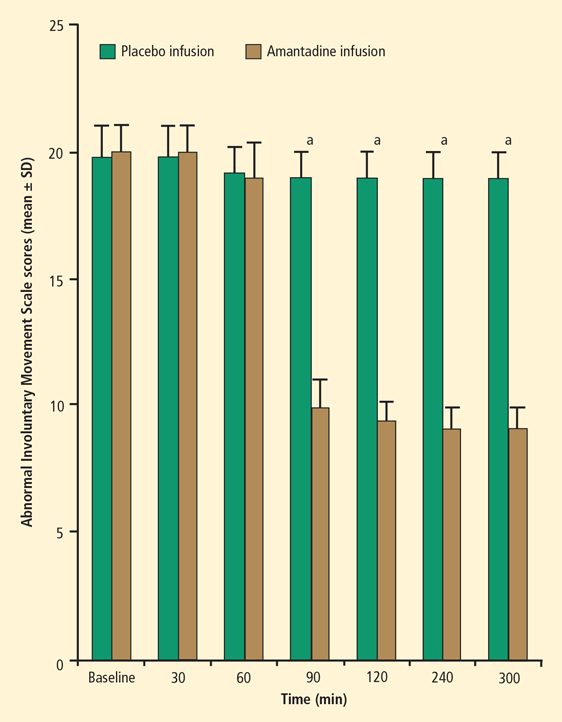Comprehensive treatment of Huntington disease and other choreic disorders





ABSTRACTThe management of choreic disorders presents significant challenges, including identifying the etiology of the disorder, treating and preventing motor symptoms, and managing a range of other neurologic and behavioral complications. Chorea may occur in several neurodegenerative, genetic, or drug-related conditions, and a thorough diagnostic evaluation is needed to identify the specific underlying causes. Some choreic disorders have specific treatable underlying etiologies, such as vitamin B12 deficiency or drug-induced dyskinesia. Autoimmune disorders such as Sydenham chorea may be treated with penicillin, corticosteroids, intravenous immunoglobulin, or plasma exchange. Heredodegenerative choreas such as Huntington disease often respond to treatment with tetrabenazine or amantadine. Many other agents may be used nonspecifically for symptom control, including benzodiazepines, neuroleptics, and antiepileptic medications. In addition to motor symptoms, patients with Huntington disease or other choreic disorders often experience increasing depression, bradykinesia, cognitive impairment, aggressive behaviors, and other complications as the disease progresses. Caring for the caregiver is also a significant concern in the long-term treatment of choreic disorders.
SYDENHAM CHOREA
Unique to Sydenham chorea is the use of penicillin as prophylaxis. Other than that, the management of Sydenham chorea exemplifies the management approach for the larger category of autoimmune choreic disorders. Pathogenic-based treatment options include immune modulation with cortico steroids, intravenous immunoglobulin (IVIG), and plasma exchange; all treatments must be administered in the appropriate clinical context.
One double-blind clinical trial examined the effectiveness of corticosteroid treatment in children with Sydenham chorea randomly assigned to receive either prednisone (n = 22) or placebo (n = 15).9 Prednisone was administered at a dose of 2 mg/kg/day for 4 weeks, followed by gradual tapering and discontinuation. The median time to remission of chorea was significantly lower for patients in the prednisone group (54.3 days) compared with those in the placebo group (119.9 days; P < .001). Patients in the prednisone group also exhibited significantly better scores on a chorea intensity rating scale at 8 weeks and 12 weeks (P < .001). Potential limitations of this approach include relapse of chorea symptoms and corticosteroid-related adverse events (eg, Cushing syndrome, hypertension).
A second study compared the effectiveness of three modalities: IVIG at a dose of 1 g/kg/day for 2 days (n = 4), plasma exchange (n = 8), and prednisone (n = 6).10 Although differences between treatment groups were not statistically significant, the authors noted that the clinical improvement in chorea symptoms tended to be greater for patients who received IVIG or plasma exchange than for those who received prednisone. Mean chorea scores improved from baseline by 72% for the IVIG group, 50% for the plasma exchange group, and 29% for the prednisone group.
After etiology-dependent treatments have been considered, several other options may be effective regardless of the specific etiology. These include symptomatic treatments such as haloperidol, atypical neuroleptics, and amantadine.11 Antiepileptic medications or benzodiazepines may also help to control symptoms, although less information is available about the use of these agents for the treatment of Sydenham chorea. Tetrabenazine may be considered for patients who will require long-term treatment.
HUNTINGTON DISEASE
Pharmacotherapy of Huntington disease may be unnecessary if symptoms are mild or not bothersome. Symptomatic treatment options include tetrabenazine, amantadine, and either first-generation neuroleptics (eg, haloperidol) or second-generation atypical neuroleptics (eg, olanzapine, quetiapine, risperidone, ziprasidone).
Treating choreas with tetrabenazine or amantadine
Considerable recent attention has focused on the efficacy and safety of tetrabenazine for the treatment of Huntington disease and other choreic disorders. Tetrabenazine is a central monoamine depleter that reversibly binds to the type-2 vesicular monoamine transporter.12 The TETRA-HD study examined the efficacy and safety of tetrabenazine for the short- and long-term control of Huntington disease.12 An initial study compared tetrabenazine with placebo in 75 patients who were treated for up to 13 weeks. In an extension study, all patients received individualized tetrabenazine doses for up to 80 weeks.
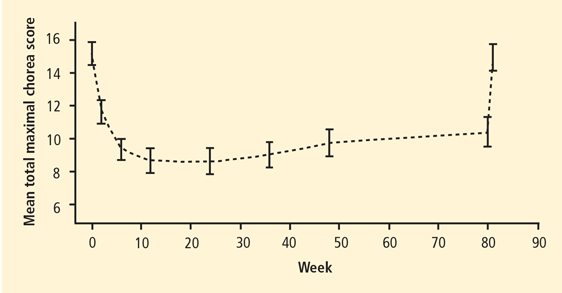
The mean total maximal chorea (TMC) scores from the Unified Huntington Disease Rating Scale (UHDRS) decreased markedly during the first 10 weeks of tetrabenazine treatment, remained lower than baseline throughout 80 weeks, and then returned to baseline levels after tetrabenazine discontinuation (Figure 1). At week 80, the mean TMC score was reduced by 4.6 UHDRS units compared with baseline (P < .001) The long-term extension phase was completed by 45 of 75 patients. Treatment-related adverse events that prompted discontinuation included depression, delusions, and vocal tics. The most commonly reported adverse events included sedation or somnolence (n = 18), depressed mood (n = 17), anxiety (n = 13), insomnia (n = 10), and akathisia (n = 9). Scores of parkinsonism and dysphagia increased significantly from baseline over the 80-week study.
Amantadine is an option for patients who cannot tolerate tetrabenazine. A double-blind, placebo-controlled study performed by researchers at the National Institutes of Health (NIH) examined the efficacy and safety of amantadine in 24 patients with Huntington disease.13 Patients were treated with oral amantadine 400 mg/day or placebo for 2 weeks, and were then crossed over to the other treatment. Amantadine was associated with a median reduction in extremity chorea score at rest of 36% from baseline (P = .04), versus 0% improvement with placebo. The mean improvement with amantadine was 56% for the 10 patients with the highest drug plasma levels.
Improvement in chorea scores from baseline for amantadine compared with placebo was rated with four different methods: (1) maximal chorea severity measured from video recordings; (2) maximal chorea severity measured by live raters; (3), chorea severity at rest measured from video recordings; and (4) extremity chorea at rest measured from video recordings. Amantadine was superior to placebo according to all four rating methods. Treatment was generally safe and well tolerated, and no consistent changes in cognitive function were noted with amantadine therapy.