Protease inhibitors: Silver bullets for chronic hepatitis C infection?





ABSTRACTRecent trials evaluated the safety and efficacy of two protease inhibitors, boceprevir (Victrelis) and telaprevir (Incivek), added to standard care with pegylated interferon and ribavirin, in patients with chronic hepatitis C virus (HCV) infection. These drugs open the door for triple therapy and other new therapies involving combinations of other direct-acting antiviral agents to become the new standard of care for this population.
KEY POINTS
- Standard care with the combination of pegylated interferon and ribavirin produces a sustained virologic response in about 40% of patients infected with HCV genotype 1, the most prevalent genotype in North America.
- New phase 3 trials showed that the addition of an oral protease inhibitor (boceprevir or telaprevir) increased the sustained virologic response rates to 70% in patients infected with HCV genotype 1.
- Boceprevir and telaprevir must be used in combination with pegylated interferon and ribavirin; they should not be used as monotherapy because of concern about the development of drug-resistant mutations.
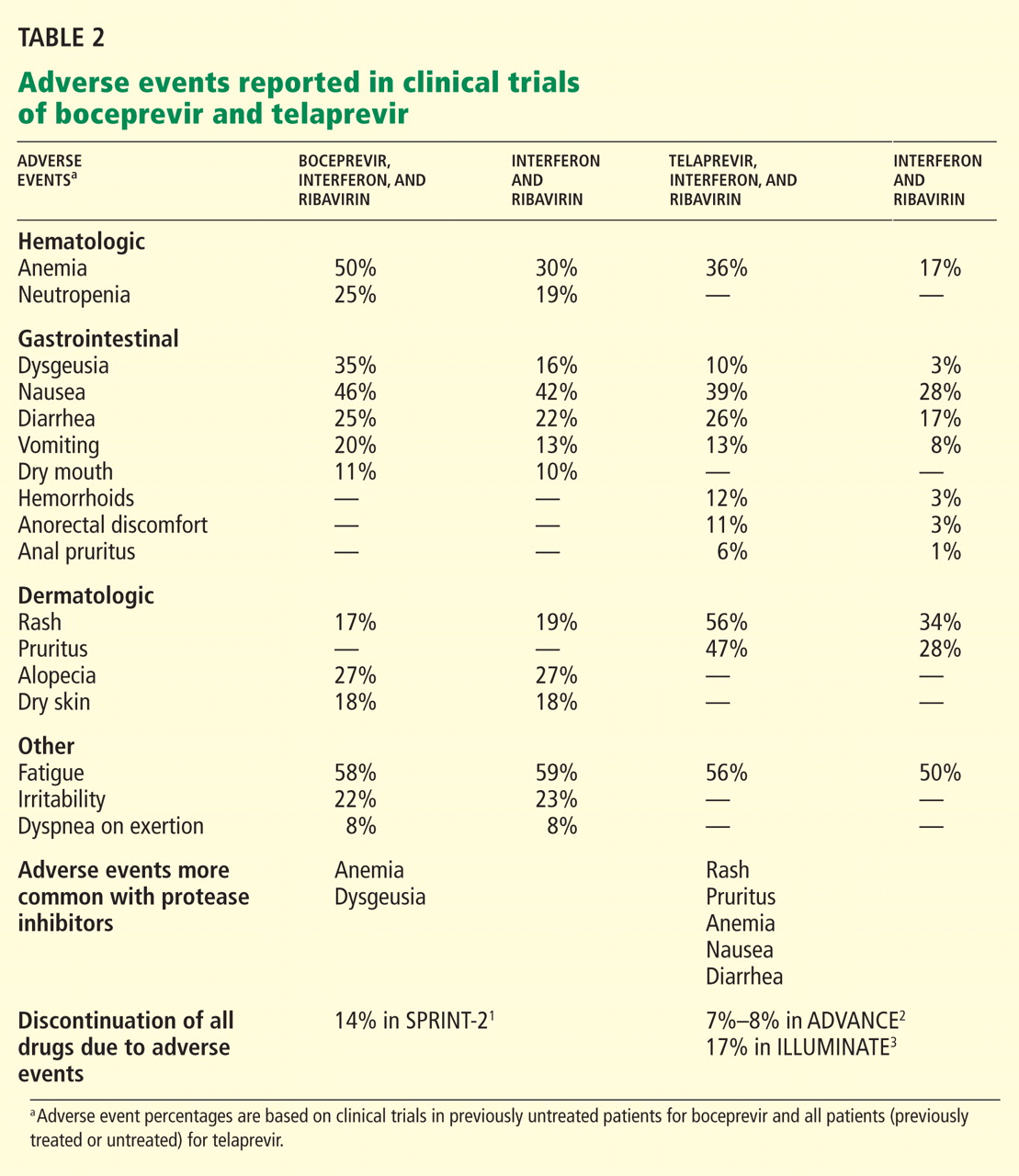
- The main side effects of boceprevir were anemia and dysgeusia. Adverse events associated with telaprevir included rash, pruritus, anemia, and diarrhea.
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.