
Detecting and managing hereditary colorectal cancer syndromes in your practice





ABSTRACTHereditary syndromes account for 5% to 10% of cases of colorectal cancer. In clinical practice, patients with these syndromes need to be identified to ensure that they and their families receive genetic counseling and testing and appropriate risk-reducing treatment. Genetic testing can offer a precise diagnosis. It allows for risk stratification and focused management and surveillance.
KEY POINTS
- Hereditary colorectal cancer syndromes carry a substantial risk of intestinal and extraintestinal tumors.
- Affected patients need increased cancer surveillance and may benefit from prophylactic surgery.
- Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history.
- Patients suspected of having hereditary colorectal cancer syndromes should be referred for genetic counseling and, if appropriate, for genetic testing.
ADENOMATOUS POLYPOSIS SYNDROMES
Familial adenomatous polyposis and MYH-associated polyposis are the next most common hereditary colorectal cancer syndromes. Each of these accounts for about 1% of cases of colorectal cancer. Clinically, these two syndromes can be challenging to distinguish because they overlap phenotypically to a significant degree.
FAMILIAL ADENOMATOUS POLYPOSIS
Familial adenomatous polyposis is caused by mutations in the APC gene. Its prevalence is 2.29 to 3.2 per 100,000 individuals.25,26
Genetics of familial adenomatous polyposis
APC is the only gene known to cause familial adenomatous polyposis. Mutations in APC are inherited in an autosomal dominant manner. Approximately 25% of cases of familial adenomatous polyposis are due to a de novo mutation in APC.27
Clinical presentation of familial adenomatous polyposis
Familial adenomatous polyposis is classified by the burden of colorectal adenomas.
Patients who have fewer than 100 adenomas have an attenuated form of the disease. In this group, polyps usually begin to form in the late teenage years or early 20s and tend to develop in the proximal colon. The attenuated form is associated with an approximately 70% lifetime risk of colorectal cancer.28
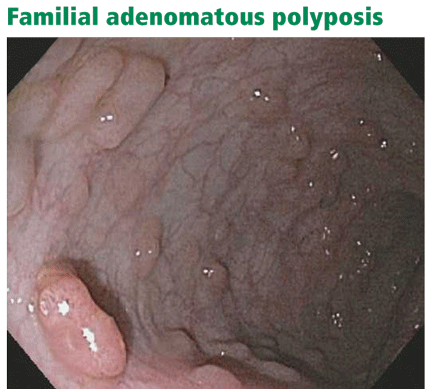
Patients who have more than 100 polyps are considered to have the classic form of the disease, and those with more than 1,000 polyps have profuse familial adenomatous polyposis (Figure 2). In these groups, polyps typically begin to develop in the preteenage to mid-teenage years. Without surgery, there is nearly a 100% risk of colorectal cancer. The average age at diagnosis of colorectal cancer is 39 years for patients with classic disease.
Upper gastrointestinal polyps are common in familial adenomatous polyposis. Nearly 90% of patients develop duodenal adenomas by a mean age of 44, with a cumulative lifetime risk of nearly 100%.29 Fundic gland polyposis occurs in nearly 90% of patients,30 while gastric adenomas are reported in fewer than 15% of patients.
Duodenal and periampullary cancer is the second most common malignancy in familial adenomatous polyposis. The lifetime risk ranges from 2% to 36%, depending on the Spigelman stage. People with Spigelman stage I, II, or III have a 2.5% risk of duodenal cancer, while those with stage IV disease have up to a 36% lifetime risk.
Gastric cancer, arising from fundic gland polyps, has been reported but is rare in Western populations.
In familial adenomatous polyposis, the incidence of jejunal adenomas and cancer is less than 10%, and the risk of ileal adenomas and cancer is less than 1%.31
Familial adenomatous polyposis is also associated with a higher risk of other malignancies, including:
- Pancreatic cancer (2% lifetime risk)
- Thyroid cancer (2% to 3% lifetime risk, typically papillary carcinoma)32
- Hepatoblastoma (1% to 2% lifetime risk)
- Brain tumors (< 1% lifetime risk)
- Biliary cancer (higher risk than in the general population).33
Benign extracolonic manifestations that have been observed include osteomas, dental abnormalities (supernumerary teeth, unerupted or absent teeth, odontomas), congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions (epidermoid cysts and fibromas), and desmoid tumors.33 The term “Gardner syndrome” has been used to describe patients who have familial adenomatous polyposis but also have osteomas and soft-tissue tumors.34 These patients carry the same risk of colorectal cancer as other patients with familial adenomatous polyposis.
Diagnosing familial adenomatous polyposis
The diagnosis of familial adenomatous polyposis is suspected when a patient has more than 10 adenomatous polyps.
Seventy-five percent of patients with familial adenomatous polyposis have a family history of the condition. Therefore, most cases are identified at a young age on screening sigmoidoscopy or colonoscopy or by predictive gene testing. Patients rarely have cancer at the time of diagnosis.
The other 25% of patients typically are diagnosed when symptoms develop from the polyps or cancer. Over 50% of these symptomatic patients have cancer at the time of diagnosis.
It is recommended that people who have more than 10 adenomas detected on a single colonoscopy or who are first-degree relatives of patients with familial adenomatous polyposis undergo a genetic evaluation and testing for mutations in the APC gene.14 Once an APC mutation is identified in the family, at-risk relatives should be offered testing around age 10 years for families with classic familial adenomatous polyposis or in the mid to late teenage years for those with the attenuated form. It also appropriate to refer patients with desmoid tumors, duodenal adenomas, and bilateral or multifocal congenital hypertrophy of the retinal pigment epithelium for a genetic evaluation.
Management of familial adenomatous polyposis
Flexible sigmoidoscopy every 1 to 2 years beginning at age 10 to 12 years is recommended for individuals and families who have been phenotypically or genetically diagnosed with familial adenomatous polyposis.35–37 If colorectal adenomas are found, surgical options should be discussed and annual colonoscopic surveillance should commence.
For people with the attenuated form, because of the later age of disease onset and the tendency for right-sided disease, colonoscopy every 1 to 2 years should commence at about age 18.35–37 If polyps are found, colonoscopy should be performed every year.
The decision of when to offer colectomy is based on polyp burden (taking into account the number, pathologic appearance, and size of the polyps) and psychosocial factors such as patient maturity. Surgical options include total colectomy and ileorectal anastomosis or total proctocolectomy and ileal pouch anal anastomosis.38 Colonic and extracolonic phenotype as well as genotype should factor into the type of operation recommended. After colectomy, annual endoscopic surveillance of the rectum or ileal pouch is indicated to screen for recurrent polyposis and cancer.
Chemoprevention with sulindac (Clinoril) 150 mg or celecoxib (Celebrex) 400 mg twice a day causes regression of colorectal adenomas in familial adenomatous polyposis and may be useful as an adjunct to endoscopy in managing the colorectal polyp burden.39,40
Forward and side-viewing upper endoscopy should commence at age 20. This should include visualization and biopsy of the papilla and periampulllary region.29 The frequency of endoscopic surveillance depends on the Spigelman stage, which reflects the duodenal polyp burden. It is recommended that patients with Spigelman stage IV duodenal polyposis be seen in consultation with an experienced gastrointestinal surgeon for consideration of a prophylactic, pylorus-preserving, pancreas-sparing duodenectomy. This procedure has been shown to be more effective in polyp control and cancer prevention than endoscopic polyp ablation and local surgical resection.41
Some evidence for the utility of celecoxib 400 mg twice daily for the regression of duodenal polyposis was noted in a 6-month placebo-controlled trial.42 Some experts recommend removal of large duodenal adenomas, with adjunctive celecoxib therapy to control polyposis burden.30
People with familial adenomatous polyposis have been shown to have a 2.6% risk of thyroid cancer, and ultrasonography of the neck with attention to the thyroid is recommended for them.32
