Pharmacovigilance and PML in the oncology setting





ABSTRACTMethods developed by the Southern Network on Adverse Reactions project, the only state-funded pharmacovigilance program in the nation, are invaluable in identifying rare and serious drug events and in disseminating related safety reports quickly throughout the medical community. An important discovery was identification and reporting of an association of rituximab and progressive multifocal leukoencephalopathy (PML) in patients without human immunodeficiency virus (HIV). A recent investigation identified 57 patients with rituximab-associated PML, including bone marrow samples, brain biopsies, and autopsy materials from patients with lymphoma and PML who tested positive for JC virus. The investigation identified an association of rituximab-chemotherapy administration and PML, although a causal relationship remains an area of active investigation. Additional investigations evaluated the epidemiology of PML in the oncology setting before and after the introduction of rituximab for lymphoma treatment. Focused analyses investigated risk factors for development of this rare complication. Further studies are needed to investigate the pathophysiology, epidemiology, and risk factors for PML developing among HIV-negative cancer patients who receive rituximab and chemotherapy.
Rare and serious drug-related events are often not detected until after clinical trials have been completed and a drug becomes widely used. Methods traditionally used by pharmaceutical companies and the US Food and Drug Administration (FDA) are not the most effective ways to promptly identify a treatment-related adverse event and quickly notify the medical community. In 1998, an academically based surveillance group was created to identify and disseminate information on unrecognized adverse drug reactions.1 In 2010, with funding from the state of South Carolina, this program became the Southern Network on Adverse Reactions (SONAR), the only state-funded pharmacovigilance initiative in the nation. SONAR and its earlier incarnation have identified potentially fatal and previously unreported side effects associated with 43 drugs—with the majority of these drugs involving the hematology and oncology discisplines.
While the earlier incarnation of drug safety monitoring relied on data mining, or detecting specific signals from large amounts of data, investigations of possible adverse drug event occurrences have a much broader scope. SONAR, an enhanced surveillance program, was created to address this issue. Based jointly at the South Carolina College of Pharmacy and a National Cancer Institute–designated cancer center, the Hollings Cancer Center at the Medical University of South Carolina, SONAR more accurately reflects the nature of our adverse effects investigations: identification of small numbers of important cases from a variety of unique data sources, including case reports, the medical literature, FDA MedWatch, and pharmaceutical manufacturers.
This article reviews methods that underlie the successful investigations of the SONAR initiative, and it examines our SONAR investigation of the association between the immune modulatory monoclonal antibody rituximab and progressive multifocal leukoencephalopathy (PML).
DETECTING, INVESTIGATING, AND DISSEMINATING FINDINGS
Surveillance programs are needed because important rare side effects are seldom discovered in a clinical trial. The Safety and Efficacy of Natalizumab in Combination with Interferon Beta-1a in Patients with Relapsing Remitting Multiple Sclerosis (SENTINEL) trial was unusual in that it detected two cases of PML associated with the use of natalizumab.2 Most rare side effects are undetected at the time of FDA approval, and usually many years elapse from the time a potential problem is detected until it is identified as a rare side effect of the drug. The average time for a “black box” warning to appear on a package insert following FDA approval is 7 to 10 years.3
Timely and thorough data collection
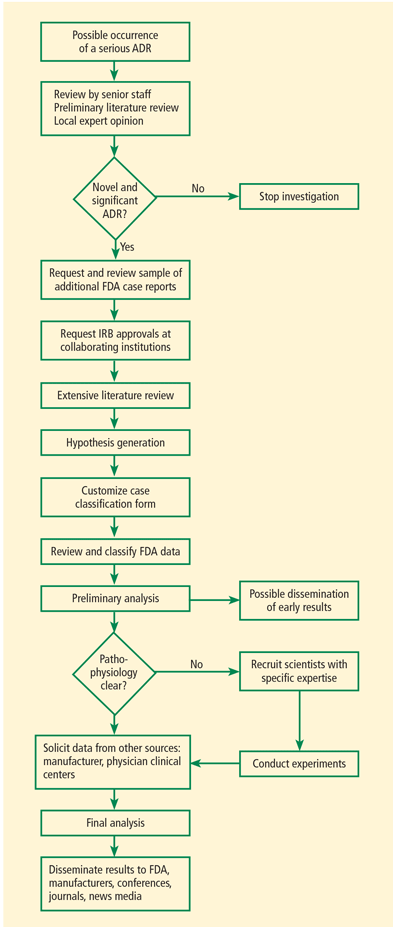
SONAR investigators perform extensive literature reviews, may request more data from authors, and request and review additional FDA case reports. Unfortunately, obtaining data from the FDA can be difficult and time-consuming. Data can be requested through the Freedom of Information Act (FOIA), but receiving it may take more than a year, and the information in the public record may be redacted. SONAR obtains laboratory tests and imaging records and works with scientists to better understand the pathophysiology of potential treatment-related rare adverse events, investigate epidemiologic estimates of the side effect rate, and evaluate risk factors for development of toxicity.
Adverse events are usually identified by SONAR within 2 years post–drug approval—a 5-year improvement over the FDA on this important metric. Once an adverse event is positively identified, the information is disseminated throughout the worldwide medical community via journal articles and presentations at medical conferences. Funding is grant-based from sources such as the National Institutes of Health (NIH), the state of South Carolina, and the University of South Carolina.
FDA, manufacturer reports may be incomplete and delayed
In contrast with SONAR, the FDA relies heavily on MedWatch to detect cases of adverse events. The safety record compiled by MedWatch is often incomplete because the program relies on voluntary submissions of adverse events; further, the inordinate amount of followup required of physicians discourages many from participating. The time to identify an adverse event can be several years, and the FDA disseminates adverse event reports via package inserts. The network that evaluates the safety information and identifies initial safety signals is mainly internal to FDA employees, as is the funding.
Pharmaceutical manufacturers frequently compile data from their own proprietary databases. Although they attempt to follow up on reports of rare adverse events, it is often difficult or impossible for the company to obtain followup information from busy clinicians. Identification of an adverse event typically takes 7 to 12 years for most pharmaceutical manufacturers—reflecting the barriers experienced in obtaining detailed information from clinicians about potential new serious adverse drug reactions. Findings are frequently disseminated through “Dear Doctor” letters. Manufacturers’ investigative networks, like those of the FDA, are largely internal and the amount of funding of they allocate to drug safety investigations is unknown.
RARE EVENTS MAY INVOLVE FEW CASES
Of our major publications,5–14 many findings are based on a small number of cases—for example, only 13 cases for clopidogrel-associated thrombotic thrombocytopenic purpura (TTP)13 and 9 for pure red cell aplasia caused by epoetin alfa.12 Important findings also come from meta-analyses,8,10 although this avenue in our pharmacovigilance approach is less typical.
The 2008 study6 on mortality and venous thromboembolism associated with erythropoiesis-stimulating agents highlights the importance of basic scientific investigation in identifying rare events. Administration of epoetin alfa to raise hemoglobin levels had been approved by the FDA in 1989 for use in patients undergoing dialysis and in 1993 for supportive use in patients with some types of cancers. We discovered that epoetin alfa promoted cancer growth based on analysis of published data and reports in conjunction with basic scientific studies of erythropoietin and erythropoietin receptors in solid cancers.