Nausea, vomiting, and panic attacks in a 50-year-old woman





IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid

Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
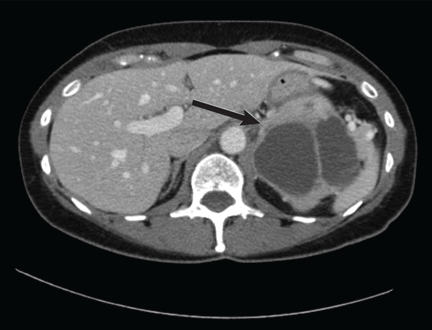
VALUE OF IMAGING STUDIES
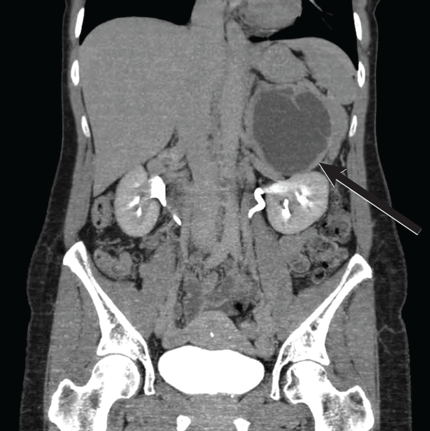
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19