Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation*





ABSTRACT
Yohimbine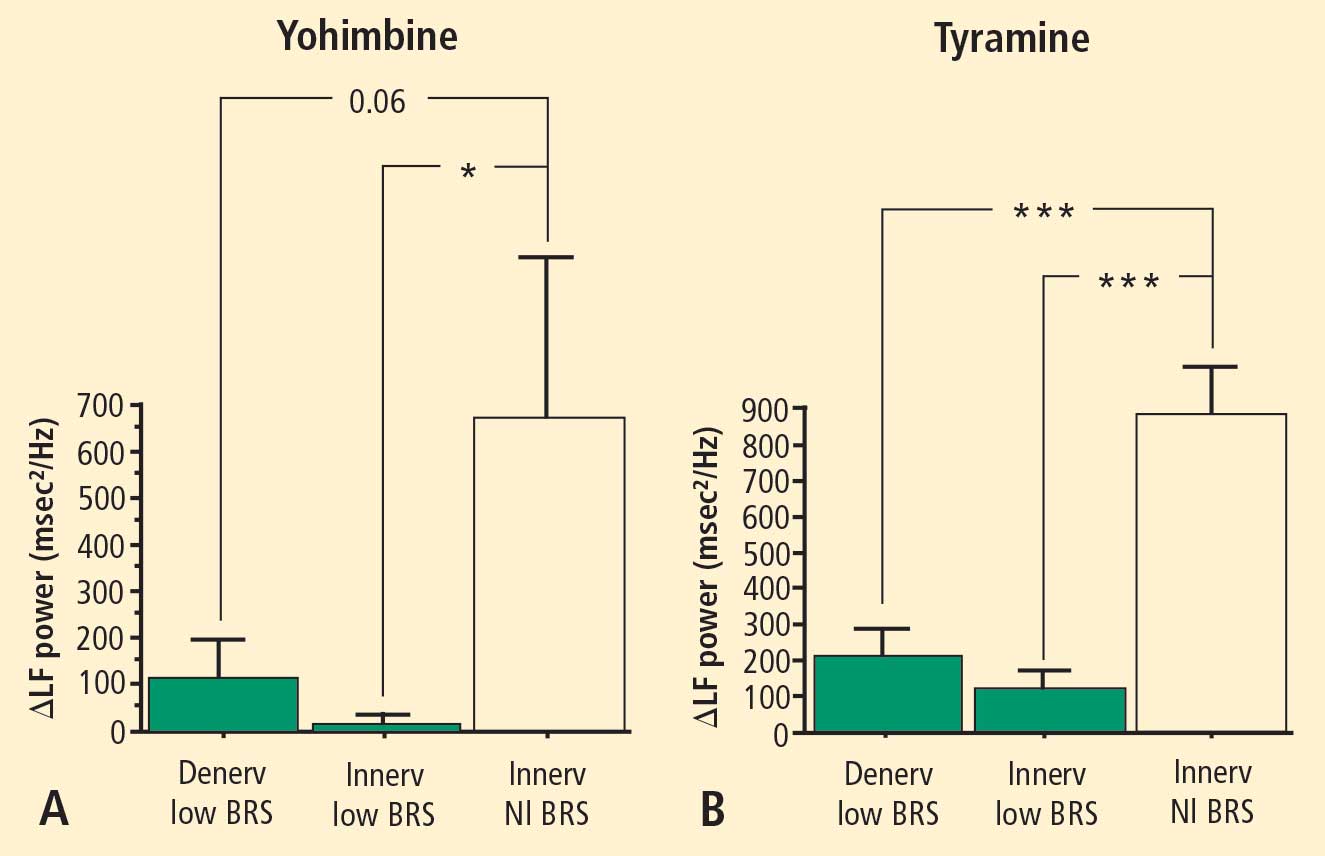
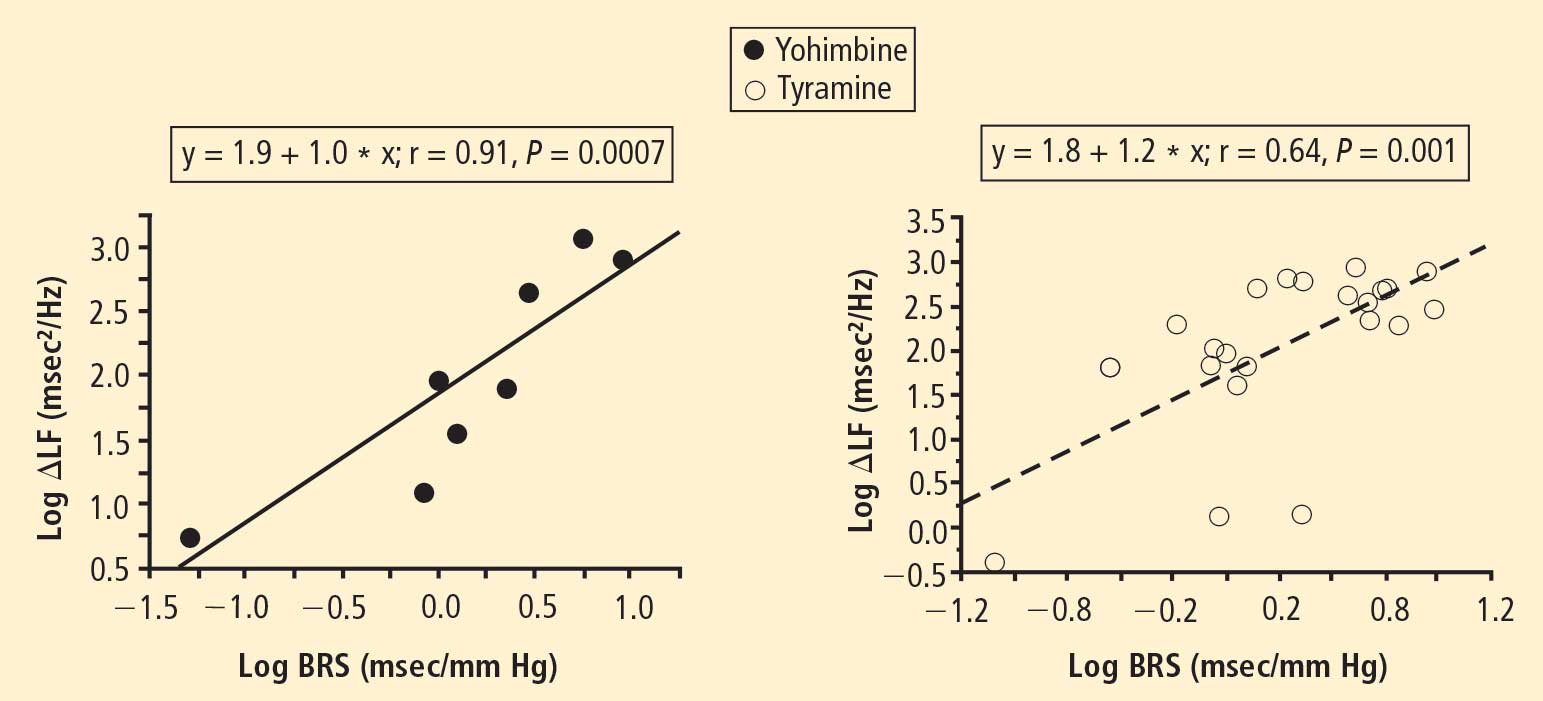
The change in LF power in response to yohimbine during cardiac catheterization was unrelated to the change in cardiac norepinephrine spillover (r = −0.09, N = 12).
Tyramine
Overall, tyramine infusion increased LF power (t = 2.9, P = 0.008). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during tyramine infusion (F = 1.7). Tyramine increased LF power in the Innervated-Normal BRS group but not in the Innervated-Low BRS or Denervated-Low BRS groups (Figure 4; data for 2 outliers excluded). The log of the change in LF power during tyramine administration was positively correlated with the log of BRS at baseline (Figure 5; data for 2 outliers excluded).
DISCUSSION
In this study, patients with neuroimaging evidence of cardiac sympathetic denervation had low LF power of heart rate variability. At first glance, this finding would seem to support the view that LF power can provide an index of cardiac sympathetic outflow. As explained below, several lines of evidence from the present study led us to infer that the association between low LF power and cardiac sympathetic innervation is determined mainly by concurrent baroreflex function.
Patients with low BRS had low LF power, and patients with normal BRS had normal LF power, regardless of the status of cardiac sympathetic innervation as assessed by 6-[18F]fluorodopamine scanning. Neither normalization of LF and HF power for total power nor use of the LF:HF ratio improved the association with indices of cardiac sympathetic innervation.
Neurochemical findings during cardiac catheterization supported the above results based on cardiac sympathetic neuroimaging. Among patients with innervated hearts who had normal cardiac norepinephrine spillover, LF power was decreased only in the group with low BRS and was normal in the group with normal BRS. As expected, cardiac norepinephrine spillover was decreased in patients with neuroimaging evidence of cardiac sympathetic denervation.
Effects of pharmacological manipulations that increase norepinephrine release from sympathetic nerves provided further support for an association between baroreflex failure and low LF power, independent of cardiac sympathetic function. Both tyramine and yohimbine increased LF power only in the subjects with normal BRS. In subjects with low BRS, neither drug increased LF power, even in the group with intact cardiac sympathetic innervation. Moreover, individual values for responses of the log of LF power to both drugs were correlated positively with the log of BRS at baseline.
The fact that HF power was positively correlated with LF power could also fit with the notion of baroreflex function acting as a common determinant of values of both variables. We cannot exclude concurrent parasympathetic cardiovagal and sympathetic denervation as an explanation for the association between HF and LF power. Inhibition of the effects of parasympathetic activity after atropine administration results in the almost complete absence of both LF and HF HRV, further suggesting a common determinant.19
Several previous investigations have cast doubt on the validity of LF power as a measure of sympathetic activity because of dissociations between LF power and cardiac norepinephrine spillover, directly recorded sympathetic nerve traffic, and plasma norepinephrine levels.4,6,20 Such dissociations are especially glaring in patients with congestive heart failure, which is characterized by decreased LF power9 despite marked cardiac sympathetic activation.3
Other pathophysiologic states do result in both decreased sympathetic nervous system activity and decreased LF power. In these pathophysiologic states, the possibility remains that low LF power might reflect failure of baroreflexive modulation of sympathetic neuronal outflows, rather than sympathoinhibition itself. For instance, Wiklund et al21 noted low LF power in patients with palmar hyperhidrosis undergoing bilateral transthoracic sympathectomy; however, baroreflex-cardiovagal sensitivity also declines after thoracic sympathectomy.22
Sleight et al8 suggested dependence of LF power on baroreflex function, based on effects of carotid baroreceptor stimulation in 3 patients: 1 with normal BRS; 1 with ischemic heart disease, congestive heart failure, and normal BRS; and 1 with ischemic heart disease, congestive heart failure, and initially low BRS who subsequently had an improved clinical state and BRS. In the baseline state, both congestive heart failure patients had low LF power, despite a presumably hypernoradrenergic state. Direct baroreceptor stimulation at 0.1 Hz increased LF power in the normal subject and in the patient with congestive heart failure and normal BRS. The congestive heart failure patient with low BRS did not have an increase in LF power until BRS normalized. These data revealed an initial dissociation between cardiac noradrenergic state in the patients with congestive heart failure and LF power. During carotid sinus stimulation, LF power increased only when BRS was normal. Low BRS obviated this effect.
Because congestive heart failure is well known to be associated with baroreflex-cardiovagal inhibition,23–25 the finding of low LF power in heart failure also supports an association between LF power and BRS, independently of increased tonic release of norepinephrine from sympathetic nerves in the heart. Cevese et al26 inhibited noradrenergic vasomotor tone using an alpha-adrenoceptor blocker in human subjects while maintaining mean blood pressure at control levels using angiotensin II. This drug combination, which would be expected to attenuate sympathetically mediated vasomotor tone and thereby decrease arterial baroreceptor input, markedly decreased or abolished LF power of HRV, suggesting that, at least under resting supine conditions, a baroreflex mechanism accounts almost entirely for LF power of HRV.
deBoer et al27 developed a beat-to-beat model of the human circulation using a set of differential equations and the following principles of operation: (1) the baroreflex regulates heart rate and peripheral vascular resistance; (2) Windkessel properties characterize the systemic arterial tree; (3) contractile properties of the ventricular myocardium follow the Starling law; and (4) respiration exerts mechanical effects on blood pressure. The model attributes LF power to a resonance in the circulatory control system, produced by a slow time constant for reflexive sympathetically mediated responses to beat-tobeat blood pressure changes. The resonance can be upregulated or downregulated by the state of baroreflex activity. The model of deBoer et al predicts that changes in blood pressure would lead heart rate changes at 0.1 Hz through a delayed sympathetic response. Changes in HR would depend on summed effects of sympathetic and vagal effects, with the sympathetic response delaying the overall response. At the respiratory frequency (0.2 to 0.3 Hz), blood pressure and HR changes would occur with little delay because of fast parasympathetic control. In essence, the response of the sympathetic nervous system behaves as a low band pass filter, with a peak response at 0.1 Hz and little response at frequencies above 0.2 Hz. Systolic blood pressure would lead to changes in heart rate via the baroreflex. In general the results of this study fit with the deBoer model.
In conclusion, LF power derived from the interbeat interval spectrogram predominantly reflects baroreflex-mediated, phasic changes in cardiovagal and sympathetic noradrenergic outflows. In the setting of baroreflex failure, baseline LF power is reduced, regardless of the status of cardiac sympathetic innervation.
LIMITATIONS
The combination of cardiac sympathetic denervation and normal baroreflex function seems quite rare. One must exercise caution in drawing inferences from the findings in the Denervated-Normal BRS group, which contained only 4 subjects, even though the difference in mean log-transformed LF power from the Denervated-Low BRS group was highly statistically significant.
All of the testing in our study was done with the subjects supine. LF power measured in other positions might have different sources and meaning.