Pioneer Award Address: Ignorance isn’t biased: Comments on receiving the Pioneer Award





ABSTRACT
Researchers ordinarily work by deriving testable hypotheses from theories using a deductive process. Hypothesis testing is inherently biased, however, because of the practical requirements of finding and publishing positive results. In contrast, ignorance isn’t biased. The combination of relevant new technology, sufficient mastery of the topic to know what is not yet known, and access to patients with rare but informative disorders sets the stage for discoveries about disease mechanisms based on induction from observations. Patient-oriented research is a strength of heart-brain medicine. Patients are a unique scientific resource because they tell us the truth. We experience the joy and thrill of a “sparkle of insight” when we realize what they teach.
This is a momentous occasion for me, for the extraordinary people in the Clinical Neurocardiology Section at the National Institutes of Health (NIH), and for my family—my wife Minka and son Joey drove all the way from Maryland late last night and early this morning to be here. I thank them publicly here.
THE ‘SPARKLE OF INSIGHT’ FROM ENLIGHTENED INDUCTION
In these brief comments, as I look back on the road I have taken over the past 40 years carrying out patient-oriented research in heart-brain medicine, I would like to convey a viewpoint instead of dwelling on the presentation of research data.
The idea I wish to convey is that ignorance isn’t biased. If you have a hypothesis you want to test, you are inherently biased to find something positive—and, if you are in academic medicine, publishable—in the data you obtain. But if you have the technical capability to measure something no one else can measure, and you have sufficient mastery of the topic to know what is not yet known, then if you make an observation that you did not predict and if you recognize its significance, you have made a discovery. You have revealed a bit of the truth. You experience the highest joy and thrill a scientist can feel—a “sparkle of insight.” When this happens, if you have sense, you stop what you have been doing to pursue that discovery.
Hardly anyone has received a Nobel Prize for testing a theory, but many Nobel Prizes have been awarded for technological advances and for discoveries based on those advances. In my view, discoverers use an enlightened inductive approach at least as much as deduction. They develop new technology that enables key novel measurements, and they keep in mind gaps in knowledge, so that they are ready to appreciate the significance of their observations.
A PERSONAL EXAMPLE
‘You have to measure something’
Let me share an example of this process by relating a sparkle of insight I had several years ago. When I began working at the NIH, I met with the chief of the Hypertension-Endocrine Branch of the National Heart, Lung, and Blood Institute about the research program I would pursue. After listening patiently to me for many minutes as I spouted about how I was going to test hypotheses derived from the concepts that people with hypertension are “hyper-tense,” and that stress causes heart disease, the chief responded, “Well, these ideas are all well and good. But what are you going to measure? You can measure whatever you want, but you have to measure something.”
Measure something. I wanted to see if there was hyperactivity of the sympathetic nervous system or excessive sympathetic innervation in hypertension, and I started working on ways to measure sympathetic activity.
The sympathetic nervous system at a glance
First I should introduce you to the sympathetic nervous system, which is one of the main effectors by which the brain regulates the heart and blood vessels. It is a key link between the brain and heart. The sympathetic nerves to the heart and other organs do not come directly from the brain but from ganglia, which are clumps of nerve cell bodies strung like pearls on a necklace on each side of the spinal column. This origin outside the central nervous system will be an important fact to keep in mind.
In the heart, the sympathetic nerves travel with the coronary arteries and then dive into the heart muscle from the outside. Sympathetic nerves also enmesh the walls of arteries and arterioles. The arterioles constitute the main determinant of total peripheral resistance to blood flow in the body and therefore figure prominently in the control of blood pressure. The architectural association between sympathetic nerves and the muscle in the heart and arteriolar walls has enticed hypertension researchers for many decades.
A false start with plasma norepinephrine measurement
I developed novel methods for measuring plasma levels of norepinephrine, which is the chemical messenger that the sympathetic nervous system uses in regulation of the circulation, and of adrenaline (epinephrine), which is the well-known and potent “fight-or-flight” hormone.1 Applying this technology to patients with high blood pressure led to several publications2–9 but actually shed more heat than light on the hypothesis of sympathetic hyperactivity as a cause of or contributor to hypertension. In the face of negative data, the theory was qualified—sympathetic hyperactivity might be apparent only in the young, or the thin, or the Caucasian, or the male—but not abandoned.
Insights from visualizing sympathetic nerves in the heart
Then I embarked on a project to visualize sympathetic nerves in the heart, by a new technology called positron emission tomographic (PET) scanning. With several colleagues—including Irwin J. Kopin, Graeme Eisenhofer, Peter Chang, David Hovevey-Zion, Ehud Grossman, and Courtney Holmes—to whom I will always be grateful, I developed a PET imaging agent called 6-[18F]fluorodopamine.10–13
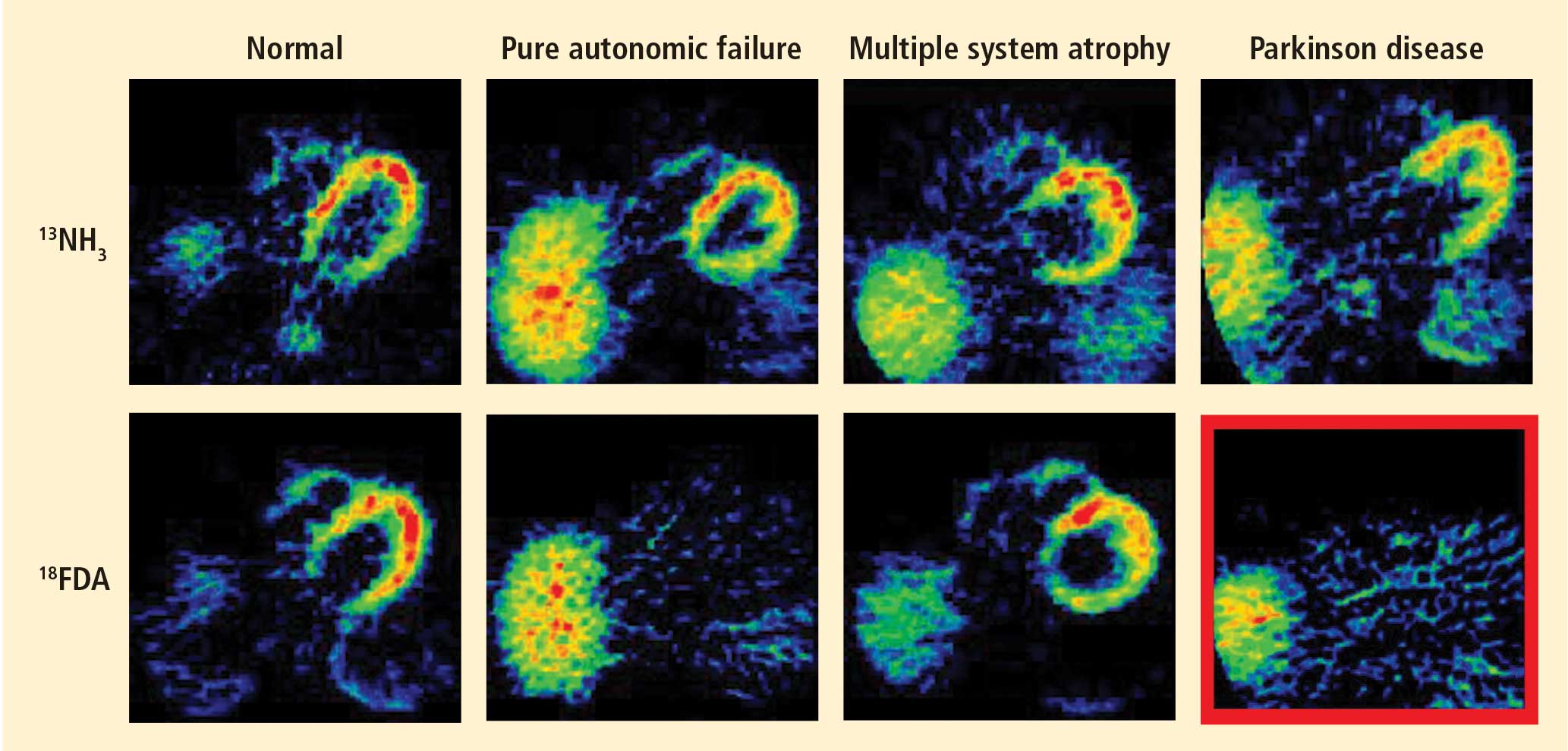
Normally, PET scans using 6-[18F]fluorodopamine look remarkably similar to scans using 13N-labeled ammonia, a perfusion imaging agent. The first patient I studied with this new technology was a patient with a rare disease called pure autonomic failure (PAF). In PAF, there was already good evidence for a loss of sympathetic nerves throughout the body. Myocardial perfusion in this patient was normal, but there was much less than normal 6-[18F]fluorodopamine-derived radioactivity in the heart muscle. In another uncommon disease, multiple system atrophy (MSA), the perfusion was also normal, and the cardiac sympathetic nerves seemed intact, in line with what was already known about this progressive neurodegenerative disease.
Then I tested a patient who had been thought to have MSA but actually had Parkinson disease (PD) with orthostatic hypotension (a fall in blood pressure each time the person stands up). PD with orthostatic hypotension can be very difficult to distinguish from the parkinsonian form of MSA. To my complete surprise, the patient with PD had a remarkable decrease in 6-[18F]fluorodopa mine-derived radioactivity in the heart muscle. There was normal blood flow to the heart muscle, so the 6-[18F]fluorodopamine was being delivered, but there was no evidence of sympathetic nerves in the heart. The scans resembled those in the PAF patient, not the MSA patient.
This finding did not arise from a prediction to test a hypothesis. It wasn’t long before I tested additional PD patients and found the same unexpected results.14,15 Because I was ignorant, I wasn’t biased. I felt I had put my finger on a piece of the truth, and I had to stop and think about the implications of this discovery. I never did come to test the hypotheses that I had sought out originally to test. Instead, I followed a totally new path, based on the discovery of cardiac sympathetic denervation in PD.