Inflammation, atherosclerosis, and arterial thrombosis: Role of the scavenger receptor CD36





ABSTRACT
The CD36 scavenger receptor recognizes oxidized low-density lipoprotein (LDL) and cell-derived microparticles. It is expressed on macrophages and platelets and is a mediator of both atherogenesis and thrombosis. Macrophages from CD36-null mice have a defect in foam cell formation in response to exposure to oxidized LDL, and CD36-null mice fed an atherogenic Western diet have significantly less atherosclerosis than their wild-type counterparts. On platelets, CD36 recognition of oxidized LDL contributes to their activation and provides a mechanistic link between hyperlipidemia, oxidant stress, and the prothrombotic state. Cell-derived microparticles are also major ligands for CD36 and contribute to thrombus formation in a CD36-dependent manner even in the absence of hyperlipidemia. CD36 deficiency in mice is associated with inhibition of thrombus formation and with a reduction in microparticle accumulation in thrombi. Targeting CD36 is a promising avenue for the treatment of atheroinflammatory disorders.
MICROPARTICLES: MAJOR LIGAND FOR CD36
Based on the experiments described above, we hypothesized the existence of endogenous CD36 ligands involved in thrombosis and proposed that cell-derived microparticles (MPs) were likely candidates. MPs are small (200–1,000 nm) phospholipid vesicles that “bud” off from cells as a result of stimulation or apoptosis. MPs can be derived from endothelial cells, leukocytes, cancer cells, and platelets; they contain selected membrane receptors as well as other proteins inherent to their parental cell (eg, MPs derived from a white cell contain tissue factor that can activate the coagulation cascade). MPs are known to circulate in patients with chronic inflammatory disorders, including acute coronary syndromes, lupus erythematosus, Wegener granulomatosis, and rheumatoid arthritis, and their number probably increases with aging.
Our hypothesis is based on the well-known observation that a major feature of MP generation is a loss of membrane asymmetry; that is, the PS normally expressed on the inner limit of the membrane instead is expressed on the surface. Previous studies from our lab and others had shown that PS and oxPS can be a ligand for CD36.11
To test our hypothesis we developed a rapid flow cytometry assay using an antibody to CD105, an antigen expressed only on endothelial cells, to detect an interaction between endothelial cell–derived MPs and platelets. This interaction is CD36-dependent in that it can be blocked with antibodies to CD36 and does not occur if platelets are taken from mice or humans who are CD36-deficient.10 MPs behaved like oxLDL in that platelets pretreated with MPs undergo a dramatic augmentation of aggregation in response to low doses of classic agonists. This augmentation of platelet activation does not occur in platelets from CD36-null donors or CD36-null mice.10
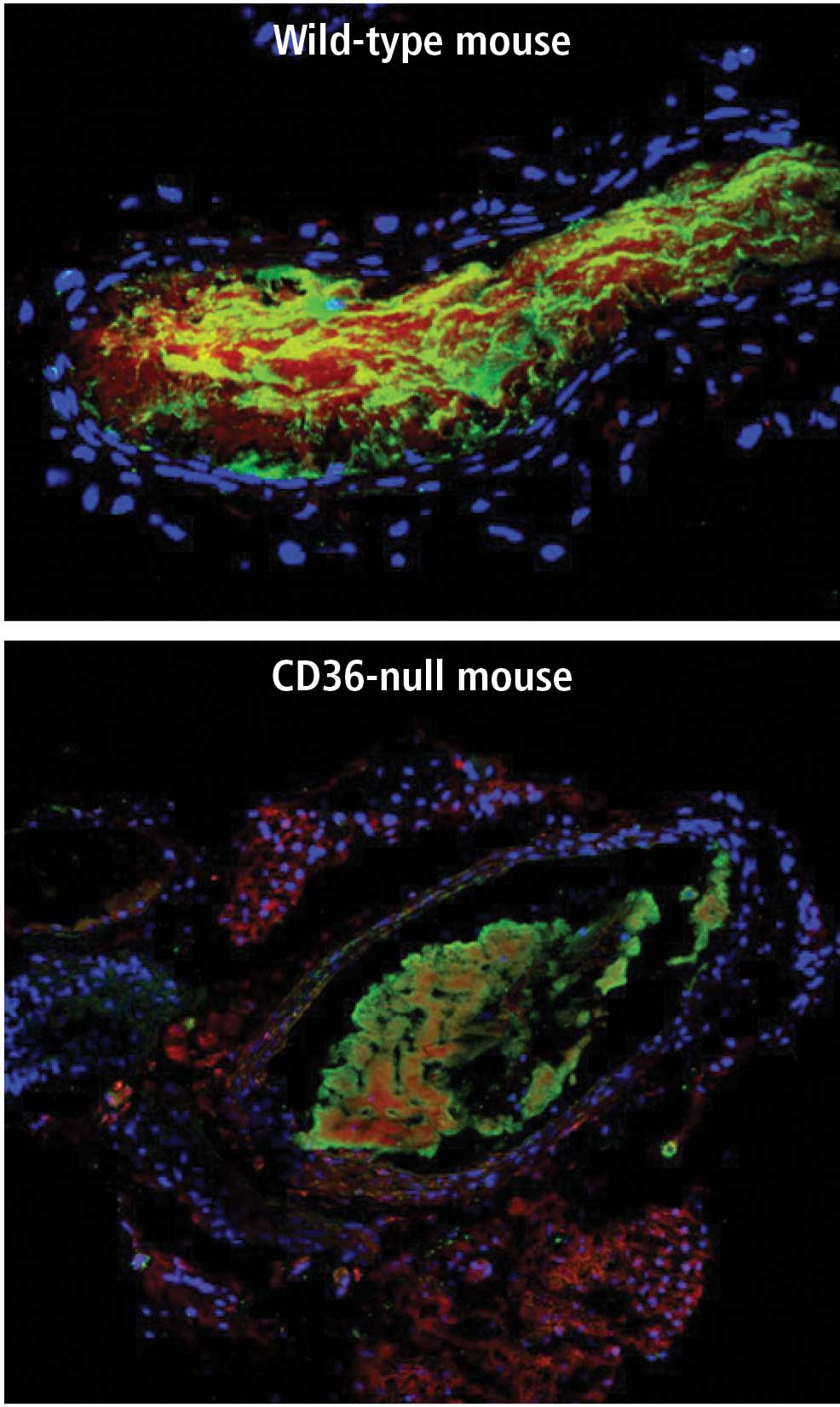
Microparticle accumulation in thrombi
Specific cytoplasmic signaling cascade
When CD36 binds to its ligands (oxLDL or MPs), it transmits a signal to the cell. In macrophages this signal leads to oxLDL internalization and foam cell formation, while in platelets it contributes to platelet activation and aggregation. In a series of studies from our laboratory and others, it has been shown that these signals are relayed by a series of molecular interactions that involve specific tyrosine kinases from the Src family and serine/threonine kinases from the mitogenactivated protein (MAP) kinase family.12 The signal to the platelet is mediated by a MAP kinase called c-Jun N-terminal kinase (JNK). Carotid thrombi in wild-type mice stain for the presence of the activated, phosphorylated form of JNK, whereas phospho-JNK expression is decreased by 50% to 60% in carotid thrombi in CD36-null mice,12 similar to the decrease in MP mass in thrombi from CD36-null mice.
CONCLUSION
These experiments suggest that CD36 has both a proatherogenic and a prothrombotic role in the vascular system. Macrophage CD36 promotes foam cell formation and plaque formation. Platelet CD36 promotes thrombosis by signaling in response to oxLDL and by phospholipids present in cell-derived MP. Therefore, targeting CD36 or CD36-signaling pathways could be a strategy in the treatment of atheroinflammatory disorders and deserves exploration.