Autosomal dominant polycystic kidney disease: Emerging concepts of pathogenesis and new treatments





ABSTRACTSome of the mystery of autosomal dominant polycystic kidney disease (ADPKD) is starting to clear. Basic research is shedding light on its pathogenesis, and new treatments are in clinical trials. This paper reviews some of these advances and what they mean to patients.
KEY POINTS
- In ADPKD the expanding cysts destroy normally functioning kidney tissue, causing hypertension, pain, and other complications, but renal function remains relatively stable until kidney volumes reach a critical size.
- Testing for genetic defects that cause ADPKD is available. The specific mutation involved (PKD1 or PKD2) affects the age of onset and therefore the rate of disease progression as well as the likelihood of cardiovascular complications. Other factors include somatic mutations (“second hits”) of the normal paired chromosome.
- Intracranial aneurysms are a key noncystic feature and may present with a very severe (“sentinel” or “thunderclap”) headache requiring immediate medical attention. Their occurrence is strongly influenced by family history.
- Basic research indicates that patients may be advised to increase their water intake, limit their sodium intake, and avoid caffeine and methylxanthine derivatives.
DOES THIS PATIENT HAVE ADPKD?
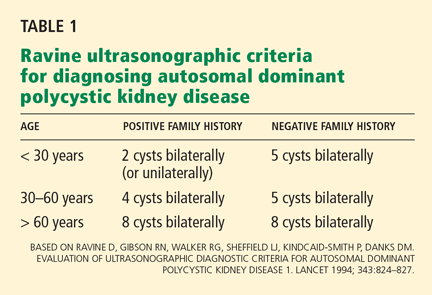
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
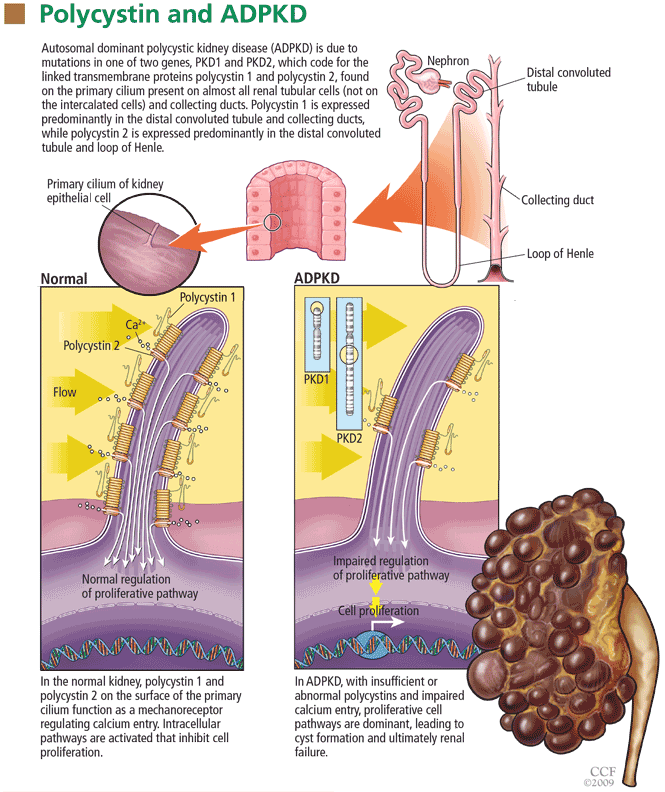
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”