An algorithm for managing warfarin resistance





ABSTRACTSome patients need higher-than-expected doses of warfarin (Coumadin) to get their international normalized ratio (INR) into the therapeutic range. The cause of warfarin resistance can be either acquired (eg, poor compliance, drug interactions, dietary interactions) or hereditary, but the genetic mechanisms of warfarin resistance are not well understood. This review offers an algorithm for the evaluation of patients with suspected warfarin resistance.
KEY POINTS
- The most common cause of warfarin resistance is noncompliance. Others include poor absorption, high vitamin K intake, hypersensitivity to vitamin K, and rapid drug deactivation.
- Patient education is necessary to improve compliance and to mitigate adverse effects of warfarin therapy, regardless of the dose.
- In time, it may be possible to individualize anticoagulant dosing on the basis of genetic testing for patients with warfarin resistance, although currently such tests are not routinely advocated and are usually done only in specialized laboratories.
- In true hereditary warfarin resistance, there are two approaches to treatment: increase the warfarin dosage (perhaps to as high as 100 mg/day or more), or switch to another anticoagulant.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
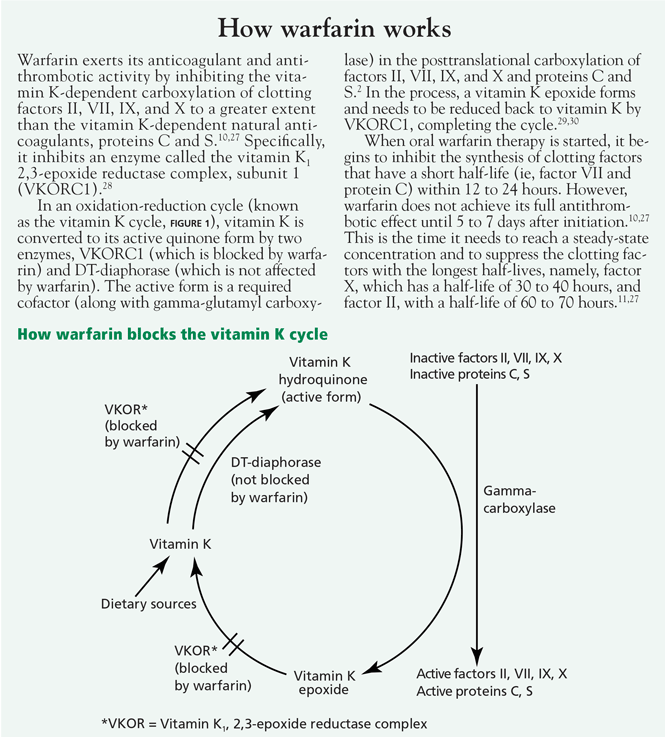
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.