Cardiovascular autonomic dysfunction in patients with movement disorders





ABSTRACT
Autonomic dysfunction is common in parkinsonian syndromes, particularly those involving dysregulation of alpha-synuclein, and may result from neurodegeneration in autonomic regulatory regions of the brain or peripherial autonomic ganglia. The most limiting cardiovascular autonomic dysfunction in these diseases is orthostatic hypotension, which is particularly prominent in multiple system atrophy. Postprandial hypotension and supine hypertension, as well as dopaminergic therapy, often complicate the management of orthostatic hypotension in patients with parkinsonian syndromes.
The Lewy body is the pathologic hallmark of both Parkinson disease and dementia with Lewy bodies. Lewy bodies are seen microscopically as neuronal inclusions containing alpha-synuclein and associated proteins. In contrast, glial inclusions involving alpha-synuclein are seen in multiple system atrophy. Because Lewy bodies are observed in autonomic regulatory regions of the brain, they are of interest in the study of the autonomic dysfunction that figures prominently in several parkinsonian syndromes. Cardiovascular autonomic dysfunction in parkinsonian syndromes includes orthostatic hypotension, postprandial hypotension, and supine hypertension.
This article will describe the major clinical and pathologic features of movement disorders with Lewy body pathology, the likelihood of autonomic dysregulation in these disorders, and issues involved in the treatment of autonomic dysfunction in patients with these movement disorders.
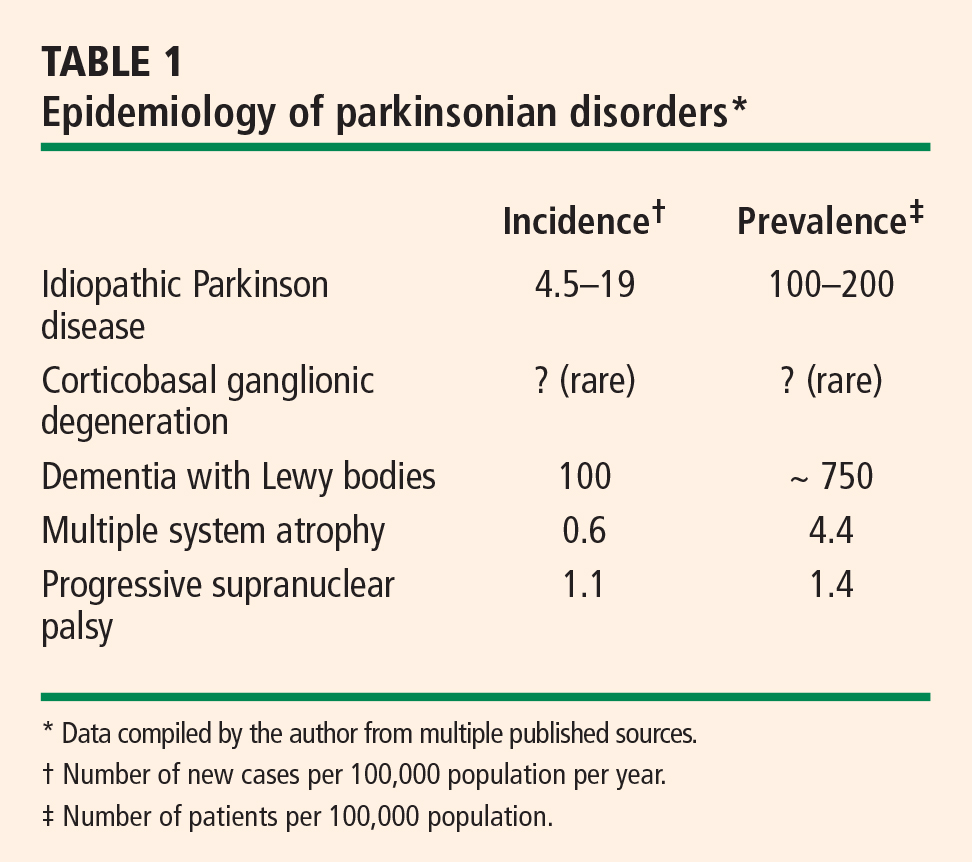
OVERVIEW OF PARKINSONIAN SYNDROMES
Tauopathies versus synucleinopathies
Pathologically, parkinsonian syndromes can be divided into two groups: the tauopathies and the synucleinopathies.
The tauopathies, so named because of the presence of hyperphosphylated tau protein, include progressive supranuclear palsy and corticobasal ganglionic degeneration as well as a number of other neurodegenerative conditions (ie, Pick disease, FTDP-17, primary progressive aphasia, argyrophilic grain disease) that do not cause parkinsonian features.
Synucleinopathies, the focus of this article, are disorders in which the protein alpha-synuclein accumulates in the cytoplasm. They include idiopathic Parkinson disease, multiple system atrophy, dementia with Lewy bodies, and pure autonomic failure. In multiple system atrophy, deposits of alpha-synuclein are prominent in glial cytoplasmic inclusions. In both Parkinson disease and dementia with Lewy bodies, alpha-synuclein is present in Lewy bodies. Although primary autonomic failure is not a movement disorder, its pathology is similar to that of the other synucleinopathies, with alpha-synuclein accumulation in both the central and peripheral nervous systems, as well as the presence of Lewy bodies.
IDIOPATHIC PARKINSON DISEASE
Autonomic dysfunction usually occurs late in idiopathic Parkinson disease, and its severity is less than that observed with other parkinsonian syndromes. However, the lifetime risk of significant autonomic dysfunction in patients with Parkinson disease is approximately 1 in 3.1 Almost 60% of patients with idiopathic Parkinson disease meet the criterion for a diagnosis of orthostatic hypotension—ie, a fall in systolic blood pressure of at least 20 mm Hg—and orthostatic hypotension is symptomatic in about 20% of patients.1
MULTIPLE SYSTEM ATROPHY
The nomenclature of multiple system atrophy has been evolving slowly. In 1900, Dejerine and Thomas described olivopontocerebellar atrophy, a progressive cerebellar degeneration with parkinsonism. In 1960, Shy and Drager described the Shy Drager syndrome, which has prominent autonomic features common to Parkinson disease, such as orthostatic hypotension, urinary and fecal incontinence, loss of sweating, iris atrophy, external ocular palsies, rigidity, tremor, loss of associated movement, and impotence.2
Also in 1960, Van der Eecken described striatonigral degeneration, an akinetic, rigid, parkinsonian syndrome that did not respond well to medications and was associated with autonomic dysfunction.3
In 1969, Graham and Oppenheimer realized an overlap to these syndromes and coined the term multiple system atrophy. They used it to refer to a gradually progressive idiopathic neurodegenerative process of adult onset characterized by varying proportions of cerebellar dysfunction, autonomic failure, and parkinsonism, and which is poorly responsive to levodopa therapy.4
Newer terminology is more specific for the predominant symptoms in the syndrome. A predominance of parkinsonism with this syndrome is referred to as “parkinsonian type of multiple system atrophy” (MSA-P), whereas a predominance of cerebellar signs is termed “multiple system atrophy with cerebellar-predominant symptoms” (MSA-C). The parkinsonian type is about four times as common as the cerebellar type.5 Autonomic dysfunction is common to both types, and its severity varies.
Parkinsonism is the most common symptom in multiple system atrophy, followed by autonomic failure and cerebellar signs. Approximately one fourth of patients with multiple system atrophy have all three categories of symptoms. Pyramidal signs are present in approximately 60% of patients, and help distinguish this syndrome primarily from idiopathic Parkinson disease.6
Diagnostic criteria
Autonomic dysfunction in the form of orthostatic hypotension and/or urinary incontinence is a key diagnostic criterion for multiple system atrophy.
Parkinsonism. Parkinsonian features of the syndrome are bradykinesia, rigidity, postural instability, and tremor.
Cerebellar dysfunction. Features of cerebellar dysfunction include gait ataxia, ataxic dysarthria, limb ataxia, and sustained gaze-evoked nystagmus.
Corticospinal tract dysfunction (extensor plantar response with hyperreflexia) also helps establish the diagnosis because this feature separates multiple system atrophy from idiopathic Parkinson disease as well as some of the other parkinsonian syndromes.
Diagnostic categories
The above diagnostic criteria can be combined to make a diagnosis of possible, probable, or definite multiple system atrophy.
Possible. For a diagnosis of possible multiple system atrophy, one of the above diagnostic criteria must be present along with two features from separate domains. If the case meets the criteria for parkinsonism (bradykinesia plus at least one of the other aforementioned features of parkinsonism), a poor levodopa response qualifies as a feature.
Probable. A diagnosis of probable multiple system atrophy must meet the criterion for autonomic dysfunction plus either the criterion for parkinsonism (with poor levodopa response) or the criterion for cerebellar dysfunction.
Definite. Definite multiple system atrophy requires pathological confirmation.
Extrapyramidal features in multiple system atrophy
In addition to prominent autonomic and/or cerebellar dysfunction, differences in extrapyramidal features help distinguish multiple system atrophy from idiopathic Parkinson disease. Tremor is less common in multiple system atrophy than in idiopathic Parkinson disease, and the akinetic/rigid symptoms tend to be symmetric in multiple system atrophy, rather than asymmetric as in Parkinson disease. Postural instability occurs early in multiple system atrophy but does not occur until late in idiopathic Parkinson disease. Moreover, multiple system atrophy responds poorly to levodopa and is characterized by more rapid disease progression. The presence of early autonomic and cerebellar symptoms is diagnostic for multiple system atrophy.
Pathology
The pathologic hallmark of multiple system atrophy is alpha-synuclein deposits in the glial or glial cytoplasmic inclusions (Papp-Lantos inclusions), which are diffuse through the central nervous system but are present particularly in the brainstem and spinal cord.