Dyspnea, arthralgias, and muscle weakness





Q: Which condition is most likely?
- Rheumatoid arthritis with pulmonary involvement
- Hypertrophic pulmonary osteoarthropathy
- Polymyositis-dermatomyositis with pulmonary involvement
- Systemic lupus erythematosus with pulmonary involvement
A: The patient’s symptoms and physical findings suggest polymyositis-dermatomyositis with associated interstitial lung disease.
Rheumatoid arthritis can also cause lung disease and proximal myopathy, but early physical findings in the hands would include symmetrical joint effusions and soft tissue swelling of the metacarpophalangeal joints.
,Patients with hypertrophic pulmonary osteoarthropathy present with arthralgias without weakness. Radiographic findings such as osteophytosis and tufting of terminal processes in the hands would support its diagnosis.
A small number of patients with systemic lupus erythematosus develop deforming arthritis with hand involvement that is either erosive (rhupus hand) or nonerosive (Jaccoud arthropathy, or lupus hand), but interstitial lung disease is rare in lupus, making this combination unlikely.
MULTIPLE PATHS TO DIAGNOSIS
Physical examination, review of systems, laboratory screening, radiographic findings, lung biopsy, electromyography, and muscle biopsy may be used in conjunction.
The criteria of Bohan and Peter are often used to diagnose polymyositis-dermatomyositis: symmetric proximal muscle weakness, elevated muscle enzymes, electromyographic changes consistent with myopathy, and compatible histologic findings on muscle biopsy, with or without the characteristic dermatologic manifestations.1,2 However, the diagnosis can be made in the typical clinical setting on the basis of characteristic levels of anti-Jo-1 antinuclear antibody and elevated serum muscle enzyme.
Depending on the criteria used, the incidence of interstitial lung disease in various studies of polymyositis-dermatomyositis ranged from 5% to 46%.3 Pulmonary involvement can present in one of three forms:
- Sudden onset of dyspnea and fever with alveolar infiltrates on chest radiography and ground-glass opacities on high-resolution chest CT
- Progressive dyspnea with radiographic findings of chronic interstitial lung disease
- No clinical symptoms, but with abnormal findings on chest radiography.4
In a minority of patients, lung disease precedes the onset of muscle or skin disease. Much more commonly, patients present with skin and muscle involvement first. In these patients, pulmonary involvement is typically seen 2 to 5 years after the diagnosis.2 Patients with interstitial lung disease are more likely to have arthralgias and arthritis than are those without lung involvement. Interestingly, the finding of microangiopathy on nail fold capillaroscopy strongly suggests pulmonary disease.3
Laboratory findings
Creatine kinase elevation is a marker of disease activity in the muscles. Aldolase, aspartate aminotransferase, and alanine aminotransferase levels may also be elevated but are not muscle-specific. Anti-Jo-1 antinuclear antibody is characteristic, although it can be negative in some patients.
Pulmonary function testing
Restrictive lung physiology with impaired diffusing capacity is the predominant pattern noted.
Lung biopsy findings
Polymyositis-dermatomyositis-associated interstitial lung disease is not limited to one histologic pattern. Nonspecific interstitial pneumonitis is the most common, but usual interstitial pneumonia, organizing pneumonia, and diffuse alveolar damage are also described.2 Patients with nonspecific interstitial pneumonitis and organizing pneumonia are suspected to have a better response to immunosuppression and better survival, although controlled studies are absent.
CT appearance
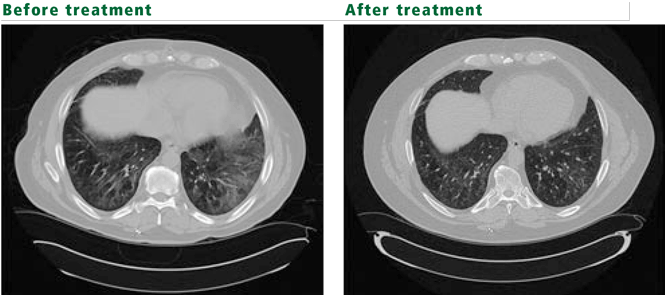
Polymyositis-dermatomyositis complicated by interstitial lung disease does not have a distinct appearance on high-resolution CT. However, the radiographic changes in most cases will suggest the underlying pathology, and this can be used to guide therapy. Most common are bibasilar subpleural ground-glass and reticular opacities that curiously spare the immediate 1 to 2 mm of subpleural parenchyma.1,3 This pattern is very suggestive of fibrotic nonspecific interstitial pneumonitis. Patchy consolidations with air bronchograms suggest organizing pneumonia. Bibasilar, subpleural honeycomb cystic changes and traction bronchiectasis are noted in usual interstitial pneumonia and suggest fibrosis, which will not improve with therapy. In patients whose disease is progressive, the areas of consolidation often evolve into honeycomb cystic changes.2,4