Preventing a first episode of esophageal variceal hemorrhage





ABSTRACTIn patients with esophageal varices, hemorrhage is common and often lethal, so we need to take a proactive approach to preventing a first episode of bleeding. All patients with cirrhosis should undergo endoscopy to look for varices. Depending on the size and appearance of the varices and the patient’s Child-Pugh grade, prophylactic treatment may be indicated.
KEY POINTS
- The hepatic vein pressure gradient (HVPG) correlates well with the portal pressure and is easier to measure. However, whether it is cost-effective to measure the HVPG in clinical practice is controversial.
- Nonselective beta-blockers are the mainstay of treatment; selective beta-blockers do not reduce portal pressure to the same degree and are not recommended for preventing variceal bleeding.
- Endoscopic variceal ligation is an acceptable alternative to beta-blocker therapy for patients who cannot tolerate these drugs and for patients with varices at high risk of bleeding.
- Nitrates are no longer used as monotherapy for preventing variceal hemorrhage, and their use in combination with beta-blockers is controversial. Surgical portal decompression, transjugular intrahepatic portosystemic shunting, and endoscopic sclerotherapy are not recommended.
Variceal hemorrhage is a medical emergency in which up to 20% of patients die.1 Even if the patient survives an initial episode of variceal bleeding, the probability of another episode is high: the rebleeding rate without treatment is 70% within 1 year. The mortality rate with rebleeding is 33%.
With such overwhelming consequences, the best strategy in any patient with cirrhosis and known varices is to try to prevent the first episode of bleeding.
WHO IS AT RISK?
Esophageal varices are present in 30% of patients with compensated cirrhosis and in up to 60% of those with decompensated cirrhosis (ie, with evidence of ascites or encephalopathy).2
The risk of variceal hemorrhage is related to three factors:
- The size of the varices. Varices 5 mm in diameter or smaller have a 7% risk of bleeding in 2 years, while those larger than 5 mm have a 30% risk of bleeding within 2 years.3
- The appearance of the varices. Morphologic features of varices, including red wale signs (red streaks of the mucosa overlying the varix), have been correlated with an increased risk of hemorrhage.
- The severity of liver dysfunction, as assessed by the Child-Pugh classification—an index of liver dysfunction based on serum albumin concentration, bilirubin level, prothrombin time, and the presence of ascites and encephalopathy. A high Child-Pugh score (ie, class B or C), representing decompensated cirrhosis, is associated with an increased risk of bleeding.
HOW VARICES DEVELOP: PORTAL HYPERTENSION
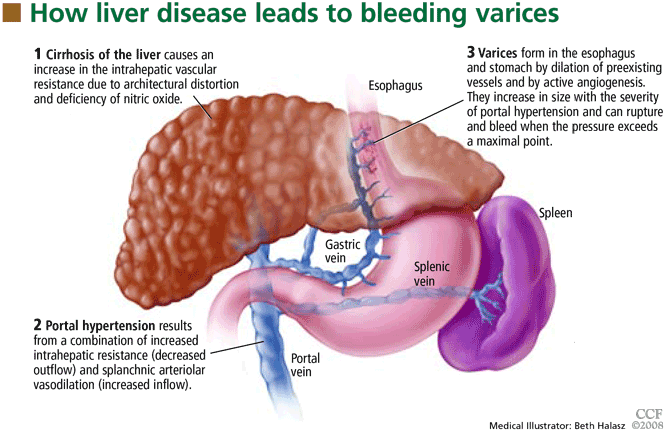
Esophageal varices form as a result of increased portal pressure, the product of increased portal venous inflow and resistance to outflow from the portal venous system. Portal hypertension is a major complication of chronic liver disease. In cirrhosis, architectural distortion of the liver causes an increase in the intrahepatic vascular resistance.
Portal venous inflow depends on mesenteric arteriolar tone, increasing when tone decreases. In cirrhotic patients, the increase in portal pressure results from a combination of increased portal blood flow secondary to splanchnic arteriolar vasodilation and elevated resistance to outflow through distorted hepatic sinusoids.
The potent vasodilator nitric oxide (NO) plays an important role in portal hypertension. In patients with cirrhosis, NO bioavailability is decreased in the intrahepatic circulation due to defects in the posttranslational regulation of endothelial NO synthase.4 This deficiency of NO, along with mechanical factors in the sinusoids, contributes to the increase in intrahepatic resistance. In the systemic and splanchnic circulation, NO bioavailability is increased due to upregulation and posttranslational regulation of endothelial NO synthase, thereby increasing splanchnic vasodilatation and leading to increased portal venous inflow.5 This results in a marked increase in cardiac output and so-called hyperdynamic circulation.
Portal hypertension results in the development of collateral circulation, including venous channels in the esophagus and stomach, by the dilation of preexisting vessels and active angiogenesis. Esophagogastric varices increase in size with the severity of portal hypertension and can rupture when the tension in their walls exceeds a maximal point.
HEPATIC VEIN PRESSURE GRADIENT: A PROXY FOR PORTAL PRESSURE
Ideally, the portal venous pressure should be directly measured. However, since direct measurement is invasive and impractical, the hepatic vein pressure gradient (HVPG) can be measured instead and correlates well with the portal pressure.6
The normal HVPG is 5 mm Hg or less; anything above this value denotes portal hypertension. However, studies have shown that varices may develop but do not bleed if the HVPG is less than 12 mm Hg.8
TWO WAYS TO PREVENT BLEEDING
Bleeding can be prevented either by reducing the portal venous pressure or by obliterating the varices. Portal pressure can be reduced by placing a portosystemic shunt either surgically or percutaneously with radiographic guidance or by giving drugs such as nonselective beta-blockers, nitrates, or a combination of these drugs. Variceal obliteration is typically done by endoscopic methods with either injection of a sclerosant or band ligation.