Screen for portopulmonary hypertension, especially in liver transplant candidates





ABSTRACTPulmonary artery hypertension may develop in some patients with liver disease and portal hypertension. Although pulmonary artery hypertension may be asymptomatic in its early stages, it should be looked for especially if a patient is a candidate for liver transplantation, as it may make transplantation riskier.
KEY POINTS
- In portopulmonary hypertension, the pulmonary artery pressures, pulmonary vascular resistance, and portal venous pressure are all elevated.
- All candidates for liver transplantation should undergo echocardiography to screen for portopulmonary hypertension. If the echocardiogram shows elevated pulmonary pressures, right heart catheterization must be performed to confirm the diagnosis.
- The ideal medical regimen remains to be determined. Although drug treatment may lower pulmonary artery pressures in selected patients so that liver transplantation can be safely done, morbidity and mortality rates remain higher in patients with moderate to severe portopulmonary hypertension.
- Liver transplantation is not the treatment of choice for portopulmonary hypertension.
VASOCONSTRICTION, REMODELING, THROMBOSIS
The pathogenesis of portopulmonary hypertension is not completely understood but likely involves a complex interaction of several mechanisms, including an imbalance of vascular mediators favoring vasoconstriction,11–13 endothelial damage with vascular remodeling due to excessive pulmonary blood flow,14,15 smooth muscle proliferation, and microvascular thrombosis.16,17
The pulmonary endothelium is a complex, dynamic organ capable of influencing a variety of vascular mediators and adapting to changes in pulmonary volume as necessary. Endothelial dysfunction may initiate the vascular changes seen in portopulmonary hypertension.
Endothelin-1 (ET-1) is a potent vasoconstrictor that has been implicated in the pathogenesis of idiopathic pulmonary artery hypertension. ET-1 levels are also increased in cirrhotic patients with refractory ascites.6
Other mediators favoring vasoconstriction include serotonin, angiotensin II, and norepinephrine. Whether these mediators influence the development of portopulmonary hypertension is not clear.
At the same time, production of vasodilatory mediators such as nitric oxide and prostacyclin may be decreased in portopulmonary hypertension, facilitating vascular remodeling and a proliferative vascular response. Prostacyclin is a potent vasodilator normally found in high concentrations in the lungs. Prostacyclin synthase is the precursor enzyme for the production of prostacyclin and is decreased in the lungs of patients with portopulmonary hypertension.18
Another way that portal hypertension may influence lung vascular tone is that endotoxin, cytokines, or both, released from the splanchnic circulation, may bypass the liver and get into the lungs.19 Evidence in support of this is that patients with portosystemic shunting can develop similar pathologic changes in the pulmonary vascular bed that normalize when the shunt is reversed. To date, however, no substance has been definitively identified.
Yet another proposed mechanism is shear stress on the pulmonary endothelium from the hyperdynamic cardiac output, with resultant vascular remodeling; however, other mechanisms must be involved, as not everyone with liver disease develops portopulmonary hypertension (see below).
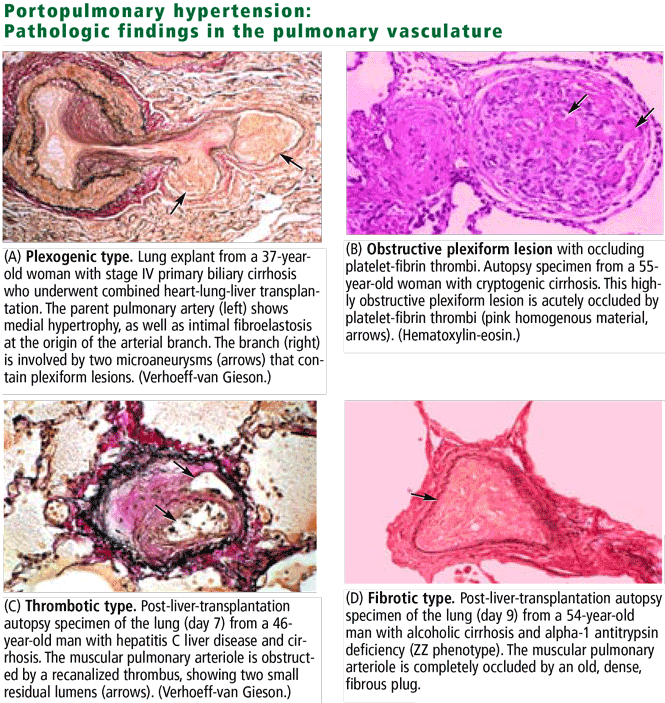
These changes are identical to those in idiopathic and familial pulmonary arterial hypertension,21 and indeed, the World Health Organization now classifies portopulmonary hypertension in the same category as these primary forms of pulmonary hypertension rather than in the secondary forms.3
Why doesn’t everyone with liver disease develop portopulmonary hypertension?
The severity of liver disease or degree of portal hypertension does not appear to correlate with the severity of pulmonary hypertension,4 and portopulmonary hypertension does not develop in all patients with portal hypertension. Therefore, it is likely that some patients have a genetic or environmental susceptibility or suffer a “second hit” that triggers dysregulated pulmonary vascular proliferation and contributes to the development of pulmonary hypertension.
Whether genetic mutations play a role in portopulmonary hypertension remains unknown. Such a mutation could be similar to the one identified in the bone morphogenetic protein receptor type 2 gene (BMPR2) in familial pulmonary artery hypertension or the mutation in the activin-like kinase gene (ALK1) seen in pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia.22
Current studies are investigating the role that bone-marrow-derived progenitor cells might play in the pathogenesis of portopulmonary hypertension.
CLINICAL FEATURES MAY NOT BE OBVIOUS AT FIRST
In the early stages of portopulmonary hypertension, patients may have no symptoms or only symptoms of liver disease, so it is important to have a high index of suspicion and screen for pulmonary hypertension. As its severity increases, symptoms may include fatigue, dyspnea, abdominal bloating, palpitations, chest pain or pressure, and syncope. The most common presenting symptom is dyspnea on exertion.
Similarly, the findings on physical examination also depend on the severity of pulmonary hypertension. Patients with mild portopulmonary hypertension may have only signs suggesting liver disease, such as spider telangiectases, jaundice, mild lower extremity edema, and ascites. As the severity of portopulmonary hypertension increases, however, findings of right heart pressure-and-volume overload become more obvious. These include peripheral edema, elevation of the jugular venous pressure, a right ventricular lift, a loud pulmonic valve closure, increased split of the second heart sound, a pulsatile liver, or a right-sided third or fourth heart sound.