Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban





ABSTRACTThe new oral anticoagulants dabigatran etexilate (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis) have predictable pharmacokinetic and pharmacodynamic profiles and are alternatives to warfarin. However, many physicians are wary of these drugs, since there is limited evidence on how to manage bleeding in patients taking them, and since no specific antidote is known to reverse their anticoagulant effect. Management requires careful adherence to first principles of bleeding care. Unapproved and untested reversal strategies may be required in patients with life-threatening bleeding.
KEY POINTS
- Thromboprophylaxis with anticoagulants is an important aspect of managing patients at risk of systemic or pulmonary embolization.
- Dabigatran is a direct inhibitor of thrombin (factor IIa); rivaroxaban and apixaban inhibit factor Xa.
- Monitoring of coagulation function is not routinely necessary with the new drugs but may be useful in emergencies.
- Nonspecific hemostatic agents that have been suggested for off-label use in reversing excessive bleeding in patients taking the new oral anticoagulants include recombinant factor VIIa, three-factor and four-factor prothrombin complex concentrate, and activated prothrombin complex concentrate.
In the past several years, three new oral anticoagulants—dabigatran etexilate (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis)—have been approved for use in the United States. These long-awaited agents are appealing because they are easy to use, do not require laboratory monitoring, and have demonstrated equivalence, or in some cases, superiority to warfarin in preventing stroke or systemic embolism in at-risk populations.1–4 However, unlike warfarin, they have no specific reversal agents. How then should one manage spontaneous bleeding problems and those due to drug overdose, and how can we quickly reverse anticoagulation if emergency surgery is needed?
For these reasons, physicians and patients have been wary of these agents. However, with a systematic approach based on an understanding of the properties of these drugs, the appropriate use and interpretation of coagulation tests, and awareness of available therapeutic strategies, physicians can more confidently provide care for patients who require urgent reversal of anticoagulant effects.
Here, we review the available literature and suggest practical strategies for management based on an understanding of the pharmacokinetic and pharmacodynamic effects of these drugs and our current knowledge of the coagulation tests.
NEED FOR ANTICOAGULANTS
Anticoagulants are important in preventing systemic embolization in patients with atrial fibrillation and preventing pulmonary embolism in patients with venous thromboembolism.
And the numbers are staggering. The estimated prevalence of atrial fibrillation in the United States was 3.03 million in 2005 and is projected to increase to 7.56 million by 2050.5 Ischemic stroke is the most serious complication of atrial fibrillation, which accounts for 23.5% of strokes in patients ages 80 through 89 according to Framingham data.6 Venous thromboembolism accounts for 900,000 incident or recurrent fatal and nonfatal events in the United States yearly.7
HOW THE NEW AGENTS BLOCK COAGULATION
Thrombin (factor IIa), a serine protease, is central to the process of clot formation during hemostasis. It activates factors V, VIII, and XI (thus generating more thrombin), catalyzes the conversion of fibrinogen to fibrin, and stimulates platelet aggregation. Its role in the final steps of the coagulation cascade has made it a target for new direct thrombin inhibitors such as dabigatran.
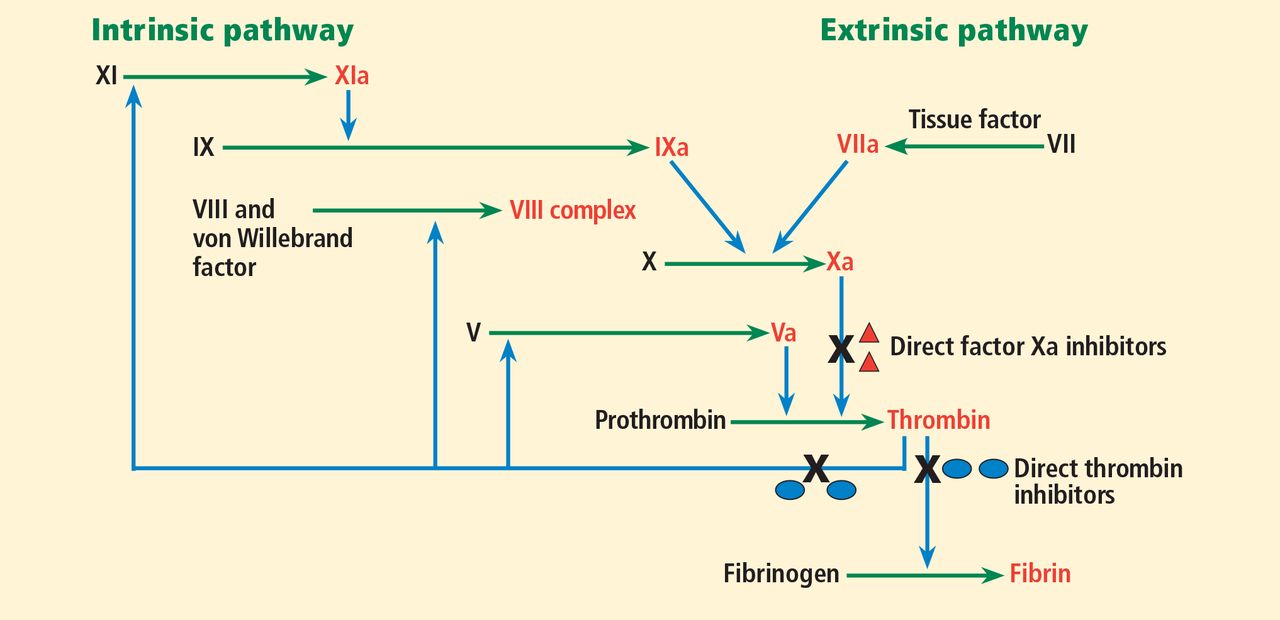
Factor Xa is a serine protease that plays a central role in the coagulation cascade. It is a desirable target for anticoagulation because it is the convergence point for the extrinsic and the intrinsic coagulation pathways. It converts prothrombin to thrombin. Rivaroxaban and apixaban are direct factor Xa inhibitors (Figure 1).
Dabigatran, a direct thrombin inhibitor
Dabigatran etexilate is a synthetic, orally available prodrug that is rapidly absorbed and converted by esterases to its active form, dabigatran, a potent direct inhibitor of both free thrombin and clot-bound thrombin.8

Plasma levels of dabigatran peak within 2 hours of administration, and its half-life is 14 to 17 hours.9 Dabigatran is eliminated mainly via the kidneys, with more that 80% of the drug excreted unchanged in the urine (Table 1).
Rivaroxaban, a factor Xa inhibitor
Rivaroxaban is a potent, selective, direct factor Xa inhibitor.
Plasma levels of rivaroxaban peak 2 to 3 hours after administration, and it is cleared with a terminal half-life of 7 to 11 hours.10,11
Rivaroxaban is eliminated by the kidneys and in the feces. The kidneys eliminate one-third of the active drug unchanged and another one-third as inactive metabolites. The remaining one-third is metabolized by the liver and then excreted in the feces. Rivaroxaban has a predictable and dose-dependent pharmacodynamic and pharmacokinetic profile that is not affected by age, sex, or body weight (Table 1).12
Apixaban, an oral factor Xa inhibitor
Apixaban is a selective, direct oral factor Xa inhibitor.
Plasma levels of apixaban peak about 3 hours after administration, and its terminal half-life is 8 to 14 hours.13 Apixaban is eliminated by oxidative metabolism, by the kidney, and in the feces. It has predictable pharmacodynamic and pharmacokinetic profiles and has the least renal dependence of the three agents (Table 1).