Advances in autosomal dominant polycystic kidney disease—2014 and beyond





ABSTRACTAutosomal dominant polycystic kidney disease (ADPKD), which frequently leads to end-stage renal disease, currently has no specific drug therapies. Better understanding of its pathogenesis and recent clinical trials have led to more accurate diagnosis of the disease and its manifestations, as well as to promising new approaches to treatment.
KEY POINTS
- For at-risk patients in the previously difficult diagnostic group from 30 to 39 years of age, newer ultrasonographic criteria for diagnosing PKD1 and PKD2 now require a minimum total of three renal cysts.
- An intracranial aneurysm occurs in approximately 16% of ADPKD patients who have a family member with ADPKD plus an intracranial aneurysm or subarachnoid hemorrhage. Appropriate screening is warranted.
- Combined positron-emission and computed tomography helps identify infected renal or liver cysts and may uncover other unsuspected abdominal or pelvic infections.
- Cyst expansion and increasing total kidney volume might be slowed by increasing water intake to 2,500 to 3,000 mL per day, although formal documentation of this is not published. However, this must be done under a physician’s supervision because of possible adverse effects.
- Tolvaptan, a promising new drug for treating ADPKD, failed to receive US approval. Rapamycin is another potentially effective agent but has had mixed results in clinical trials.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disease, has an estimated prevalence of 1:400 to 1:1,000 live births in the United States, and occurs worldwide.1,2 There are about 700,000 people living with it in the United States, and about 6,000 new cases arise annually. It accounts for nearly 5% of all patients with end-stage renal disease in the United States.3
This paper will offer an overview of the pathogenesis of renal cysts, review some of the clinical aspects of ADPKD including diagnosis and management of complications, and discuss recent drug trials and current management.
TWO TYPES—PKD1 IS MORE COMMON AND PROGRESSES MORE RAPIDLY
Two major forms of ADPKD are recognized and can usually be determined by genetic testing: PKD1, accounting for about 85% of cases, and PKD2, accounting for 15%.
The gene locus for PKD1 is on the short arm of the 16th chromosome (16p13.3), and its glycoprotein gene product is polycystin 1 (PC1), a large molecule with 4,303 amino acids.2 PC1 has a long N-terminal extracellular tail that can function as a mechanosensor. Disease progression is much faster with PKD1, and end-stage renal disease usually occurs before age 56.4
In PKD2, the gene locus is on the long arm of the fourth chromosome (4q21–23), and has a smaller glycoprotein gene product, polycystin 2 (PC2), that plays a role in calcium transport. The disease course of PKD2 tends to be slower. End-stage renal disease might not develop in the patient’s lifetime, since it typically develops when the patient is more than 70 years old.4
Although the growth rate of renal cysts is similar between the two types, patients with PKD1 develop about twice as many cysts as those with PDK2, and their cyst development starts at a younger age.5
Typically, patients have a clear phenotype and a positive family history, but in about 10% of possible ADPKD cases, there is no family history of ADPKD. Genetic variations such as incompletely penetrant PKD1 alleles,6 hypomorphic alleles,7 and trans-heterozygous mutations8 account for at least some of these cases.
IMAGING CRITERIA HAVE BROADENED
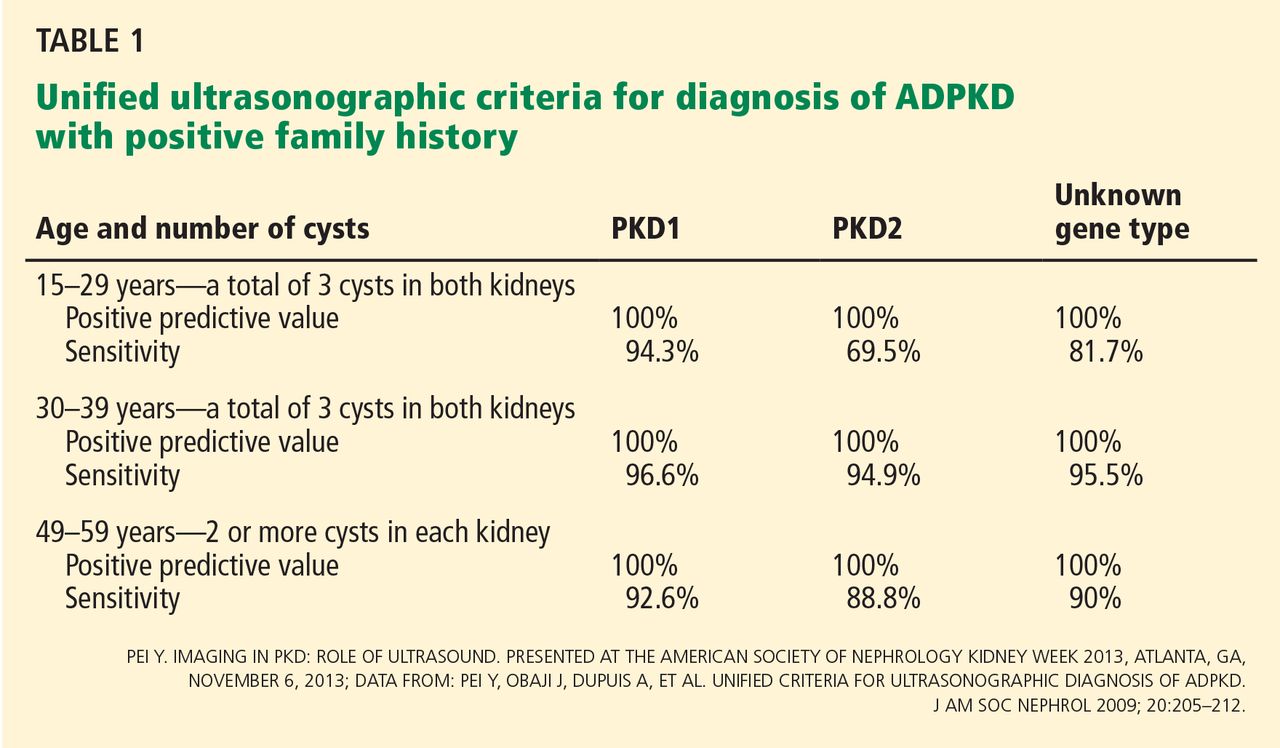
Ultrasonographic criteria for the diagnosis of ADPKD that were published in 1994 were based on patients who had a family history of PKD1.9 The criteria have since been modified (the “unified criteria”) to include patients with a family history of PKD2 who begin cyst development at a later age and with lower numbers.10 For patients ages 30 to 39, a previously difficult diagnostic group, the criterion for the minimum number of cysts visible on ultrasonography changed from four to three, improving the sensitivity of detecting disease from approximately 76% to approximately 95% (Table 1).9,10 It is important to note that these criteria apply only to patients “at risk,” ie, with a positive family history of ADPKD.
Computed tomography (CT) and magnetic resonance imaging (MRI) classically show bilaterally enlarged multicystic kidneys, though variations can be seen.
DISEASE CAN PRESENT IN MYRIAD WAYS

Although cystic kidney disease is the basic underlying problem, undiagnosed patients may present with a variety of symptoms caused by other manifestations of ADPKD (Table 2).
Hypertension is the most common presentation, occurring in about 50% of patients ages 20 to 34, and essentially 100% of those with end-stage renal disease.11 It is associated with up-regulation of the renin-angiotensin-aldosterone system.
Pain is typically located in the abdomen, flank, or back and can occur in a localized or diffuse manner. Early abdominal distress is often simply described as “fullness.” Localized pain is usually caused by bleeding into or rupture of a cyst, renal stones, or infection.12 Because renal cysts are noncommunicating, bleeding can occur into a cyst and cause pain without gross hematuria. Compression by greatly enlarged kidneys, liver, or both can cause a variety of gastrointestinal symptoms such as reflux esophagitis and varying degrees of constipation. Diffuse pain is often musculoskeletal and related to exaggerated lordosis from increasing abdominal size due to enlarging cystic kidneys and sometimes liver.12 In carefully selected cases, cyst aspiration may be helpful.11
Although renal carcinomas are rare and not more frequent than in the general population, they can occur at an earlier age and with constitutional symptoms.11
Urinary tract infections are increased in frequency. A patient may have a simple urinary tract infection that is cured with the appropriate antibiotic. However, a urinary tract infection repeatedly recurring with the same organism is a strong clue that an infected cyst is the source and requires more intensive treatment with the appropriate cyst-penetrating antibiotic. On the other hand, because cysts are noncommunicating, an infected cyst might be present despite a negative urine culture.
Identifying infected cysts can be a challenge with conventional imaging techniques, but combined positron emission tomography and CT (PET/CT) can be a valuable though expensive diagnostic tool to identify an infected kidney or liver cyst, or to identify an unsuspected source of the pain and infection.13
Jouret et al13 evaluated 27 PET/CT scans performed in 24 patients with ADPKD and suspicion of an abdominal infection. Patients were deemed to have probable cyst infection if they met all of the following criteria: temperature more than 38°C for longer than 3 days, loin or liver tenderness, plasma C-reactive protein level greater than 5 mg/dL, and no evidence of intracystic bleeding on CT. Patients with only two or three of these criteria were classified as having fever of unknown origin. Diagnosis of cyst infection was confirmed by cyst fluid analysis.
PET/CT identified a kidney or liver cyst infection in 85% of 13 infectious events in 11 patients who met all the criteria for probable cyst infection; CT alone contributed to the diagnosis in only one patient.13 In those with fever of unknown origin, PET/CT identified a source of infection in 64% of 14 events in 13 patients: two infected renal cysts, as well as one patient each with other infections that would be difficult to diagnose clinically, ie, small bowel diverticulitis, psoas abscess, diverticulitis of the right colon, pyelonephritis in a transplanted kidney, infected abdominal aortic aneurysm, prostatitis, colitis, and Helicobacter pylori gastritis. Results of PET/CT were negative in five patients with intracystic bleeding.
Kidney stones occur in 20% to 36% of patients.11,14 Uric acid stones occur at almost the same frequency as calcium oxalate stones.
Chronic kidney disease not previously diagnosed may be the presenting condition in a small percentage of patients, sometimes those in whom much earlier hypertension was not fully evaluated. ADPKD is typically not associated with significant proteinuria (eg, nephrotic range), and the presence of heavy proteinuria usually indicates the presence of a superimposed primary glomerulopathy.15
Cysts in other locations. By MRI, liver cysts are present in 58% of patients ages 15 to 24, rising to 94% in those ages 35 to 46.11 Because liver cysts are estrogen-dependent, they are more prominent in women. A small percentage of patients develop cysts in the pancreas (5%), arachnoid membranes (8%), and seminal vesicles (40% of men with ADPKD).11
Cardiovascular abnormalities occur in almost one-third of patients with ADPKD, usually as mitral and aortic valve abnormalities.16 Aneurysms of the aortic root and abdominal aorta can also occur, in addition to intracranial aneurysms (see below).17
Intracranial aneurysms are not uncommon, and size usually determines their risk.
Intracranial aneurysms are strongly influenced by family history: 16% of ADPKD patients with a family history of intracranial aneurysm also develop them, compared with 5% to 6% of patients with no family history.11 The anterior cerebral circulation is involved in about 80% of cases. A sentinel or sudden “thunderclap” headache is a classic presentation that may precede full-blown rupture in about 17% of cases.18 Patients who rupture an intracranial aneurysm have a mean age of 39, usually have normal renal function, and can be normotensive.11
For patients with no history of subarachnoid hemorrhage, the 5-year cumulative rupture rates for patients with aneurysms located in the internal carotid artery, anterior communicating or anterior cerebral artery, or middle cerebral artery were 0% for aneurysms less than 7 mm, 2.6% for those 7 to 12 mm, 14.5% for those 13 to 24 mm, and 40% for those 25 mm or larger, with higher rates for the same sizes in the posterior circulation.11
In patients without symptoms, size is correlated with risk of rupture: less than 4 mm is usually associated with very low risk, 4 to less than 7 mm with moderate risk, and 7 mm or more with increasing risk. An aneurysm larger than 10 mm is associated with roughly a 1% risk of rupture per year.19
Irazabal et al20 retrospectively studied 407 patients with ADPKD who were screened for intracranial aneurysm. Saccular aneurysms were detected in 45 patients; most were small (median diameter 3.5 mm). During cumulative imaging follow-up of 243 years, only one new intracranial aneurysm was detected (increasing from 2 to 4.4 mm over 144 months) and two previously identified aneurysms grew (one increasing 4.5 to 5.9 mm over 69 months and the other 4.7 to 6.2 mm over 184 months). No change occurred in 28 patients. Seven patients were lost to follow-up, however. During cumulative clinical follow-up of 316 years, no aneurysm ruptured. Two patients were lost to follow-up, three had surgical clipping, and five died of unrelated causes. The authors concluded that presymptomatic intracranial aneurysms are usually small, and that growth and rupture risks are no higher than for unruptured intracranial aneurysms in the general population. A 2014 study also suggests a conservative approach for managing intracranial aneurysm in the general population.21
In asymptomatic ADPKD patients, it is reasonable to reserve screening for those with a positive family history of intracranial aneurysm or subarachnoid hemorrhage, those with a previous ruptured aneurysm, those in high-risk professions (eg, pilots), and for patients prior to anticoagulant therapy or major surgery possibly associated with hemodynamic instability.11,22 Certain extremely anxious patients might also need to be studied. Screening can be performed with magnetic resonance angiography without gadolinium contrast. It is prudent to have patients with an intracranial aneurysm thoroughly evaluated by an experienced neurosurgeon with continued follow-up.