Importance of platelets and platelet response in acute coronary syndromes





ABSTRACT
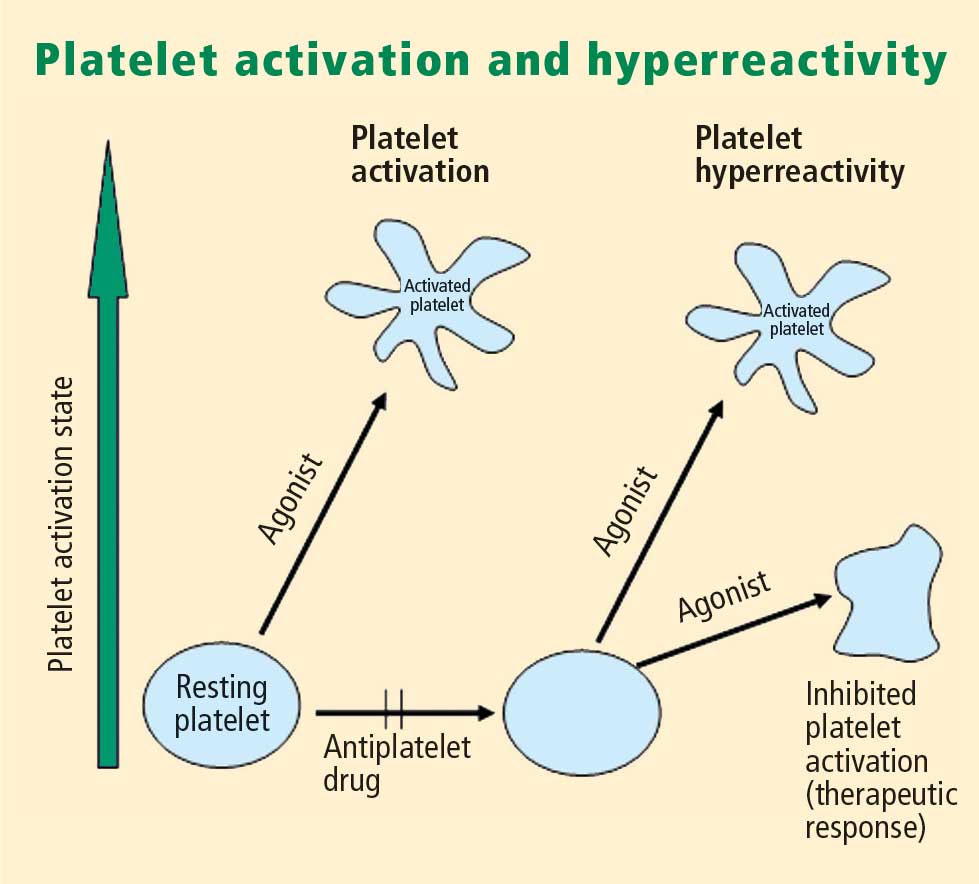
Platelet activation is one of the essential steps in the genesis and propagation of atherothrombosis. Accumulating clinical evidence suggests that an elevated platelet count, platelet activation, and platelet hyperreactivity (defined as residual platelet activity despite antiplatelet drug therapy) may be associated with adverse cardiovascular events in patients with acute coronary syndromes. Platelet function can be analyzed using various assays and measures of platelet activation. The best assays for measuring residual platelet activity in the setting of antiplatelet therapy are still being defined, as are their predictive values. Platelet aggregation remains the gold standard, but other testing methods offer advantages for specific applications, such as detecting overall platelet hyperreactivity in the presence of antiplatelet therapy or detecting inhibition of the adenosine diphosphate receptor P2Y12. Standard testing protocols for platelet aggregation are needed to achieve consistency among studies.
KEY POINTS
- Platelet function assays are inherently variable because they measure cell function rather than a single analyte.
- Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of different aspects of platelet function.
- There appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major cardiac events associated with platelet hyperreactivity.
- For predicting cardiac events, receiver operating characteristic (ROC) curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrary cutoff values.
PLATELET FUNCTION
Platelets are non-nucleated cells produced by megakaryocytes, which are very large cells (50 to 100 μm in diameter) found in bone marrow. The megakaryocyte surface membrane forms protoplatelet extensions from which platelets “bud off” and are emitted into the circulation, where they number approximately 200,000 to 400,000 per microliter of blood.
Platelet activation
Platelets play a crucial role in the vascular response to injury, and activation of platelets has long been recognized as an important step. Platelets release dense granules that contain the nucleotide adenosine diphosphate (ADP), which activates other platelets. They also possess alpha granules, which contain proteins and protein mediators (eg, platelet-derived growth factor, platelet factor 4) that are involved in inflammatory processes. The platelet surface is coated with hundreds of thousands of receptors for other cells, including activated vascular wall cells and extracellular matrix proteins. Platelets possess an affinity for adherence, especially to injured vessel walls, where they release their granule contents and then aggregate. These properties promote platelets’ involvement in many vascular processes, including ACS, as will be explored below.
Platelets exist in a nonactivated state and are drawn passively into areas of vascular injury. Initially, they adhere to proteins such as von Willebrand factor, which is a large extracellular matrix protein produced by endothelial cells. The platelet glycoprotein Ib/IX/V binds to von Willebrand factor, forming a loose association that results in platelets rolling on the surface of the vessel wall. As a multimer, von Willebrand factor exists in one subunit that is dimerized and then polymerized, making it an ideal substrate for platelets because of the multiple substrates to which platelets can adhere.
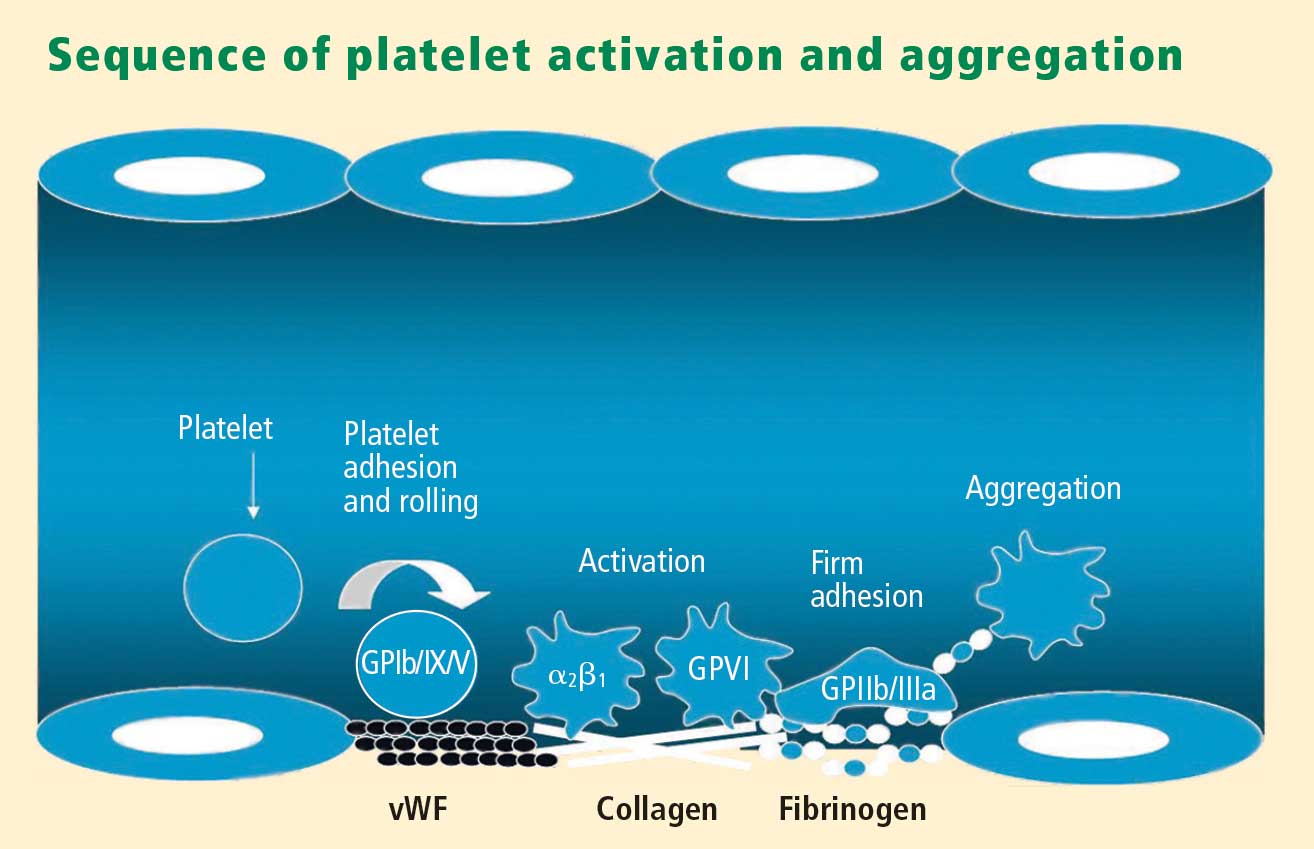
Platelet fibrinogen receptor
The platelet fibrinogen receptor (glycoprotein IIb/IIIa receptor) is an αIIbβ3 integrin that binds to arginine-glycine-aspartic acid (RGD) epitopes of proteins, such as fibrinogen. Fibrinogen has a two-dimensional symmetry, with RGD groups on both ends of the molecule, which makes it an ideal molecule for linking platelet to platelet.
von Willebrand factor has RGD groups, as do both fibronectin and glycoprotein IIb/IIIa vitronectin, and can therefore bind to many plasma and extracellular matrix proteins. The glycoprotein IIb/IIIa receptor is inactive in resting platelets. It becomes activated during the platelet activation process and binds to fibrinogen, which bridges to other platelets, causing aggregation.
ADP receptors
Various receptors on platelet surfaces are responsible for platelet activation. One is a family of receptors for ADP. As ADP is released from platelets, it can then activate other platelets by binding to the receptors. The ADP receptor P2Y12 signals through G protein pathways and is coupled to adenylate cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate to cyclic adenosine monophosphate (cAMP). High levels of cAMP inhibit platelet function; ADP binding to P2Y12 shuts down adenylate cyclase, which leads to phosphoinositide 3-kinase activation and accelerated aggregation and platelet release.
A final notable factor in the mediation of platelet activation and aggregation is phospholipase A2, which liberates arachidonic acid from the platelet membrane, metabolizing it through cyclooxygenase and thromboxane synthase to generate thromoboxane A2, which leads to release of platelet granule contents and aggregation of other platelets.