Left ventricular thrombosis can still complicate acute myocardial infarction





A 62-year-old man with hypertension, type 2 diabetes mellitus, and hypercholesterolemia presented to the emergency department with substernal chest pain that started about 15 hours earlier while he was at rest watching television.
On examination, his pulse was 92 beats per minute and regular, his blood pressure was 160/88 mm Hg, and he had no evidence of jugular venous distention or pedal edema. Lung examination was positive for bibasilar crackles.
Electrocardiography revealed Q waves with ST elevation in leads I, aVL, V4, V5, and V6 with reciprocal ST depression in leads II, III, and aVF.
His troponin T level on presentation was markedly elevated.

He underwent heart catheterization and was found to have 100% occlusion of the proximal left anterior descending artery. He underwent successful percutaneous coronary intervention with placement of a drug-eluting stent, and afterward had grade 3 flow on the Thrombolysis in Myocardial Infarction (TIMI) scale.
Echocardiography the next day revealed a mobile echo-dense mass in the left ventricular apex (Figure 1) and a left ventricular ejection fraction of 35%.
THE INCIDENCE OF LEFT VENTRICULAR THROMBOSIS IN ACUTE MI
1. What is the incidence of left ventricular thrombosis after acute myocardial infarction (MI), now that primary percutaneous coronary intervention is common?
- 0.1%
- 2%
- 20%
- 40%
Left ventricular thrombosis is a serious complication of acute MI that can cause systemic thromboembolism, including stroke.1 Before thrombolytic therapy was available, this complication occurred in 20% to 60% of patients with acute MI.2,3 But early reperfusion strategies, anticoagulation for the first 48 hours, and dual antiplatelet therapy have reduced the incidence of this complication significantly.
In the thrombolytic era, the incidence of left ventricular thrombosis was 5.1% in the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI) 3 study, which had 8,326 patients. A subset of patients who had an anterior MI had almost double the incidence (11.5%).3

The incidence has further declined with the advent of primary percutaneous coronary intervention, likely thanks to enhanced myocardial salvage, and now ranges from 2.5% to 15% (Table 1).4–11 The largest observational study, with 2,911 patients undergoing percutaneous coronary intervention, reported an incidence of 2.5% within 3 to 5 days of the MI.7 At our center, the incidence was found to be even lower, 1.8% in 1,700 patients presenting with ST-elevation MI undergoing primary percutaneous coronary intervention. Hence, of the answers to the question above, 2% would be closest.
Large infarct size with a low left ventricular ejection fraction (< 40%), anterior wall MI, hypertension, and delay in time from symptom onset to intervention were independent predictors of left ventricular thrombus formation in most studies.7,12 The risk is highest during the first 2 weeks after MI, and thrombosis almost never occurs more than 3 months after the index event.5,13–16
WHAT IS THE PATHOGENESIS OF LEFT VENTRICULAR THROMBOSIS?
A large transmural infarct results in loss of contractile function, which causes stagnation and pooling of blood adjacent to the infarcted ventricular segment. In addition, endocardial injury exposes tissue factor, which then initiates the coagulation cascade. To make matters worse, MI results in a hypercoagulable state through unclear mechanisms, which completes the Virchow triad for thrombus formation. Elevations of D-dimer, fibrinogen, anticardiolipin antibodies (IgM and IgG), and tissue factor have also been reported after acute MI.17
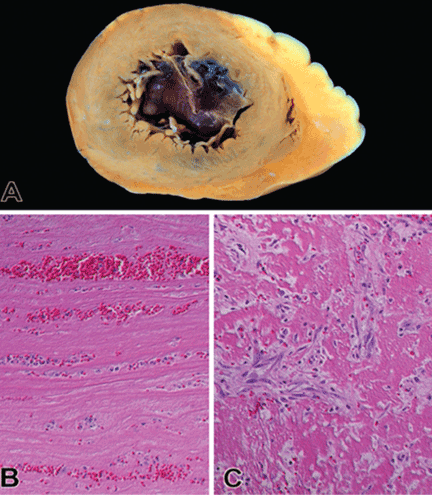
Thrombus formation begins with platelet aggregation at the site of endocardial damage, forming a platelet plug, followed by activation of clotting factors. These thrombi are referred to as “mural,” as they adhere to the chamber wall (endocardium). They are composed of fibrin and entrapped red and white blood cells (Figure 2).
The natural course of thrombus evolution is established but variable. A left ventricular thrombus may dislodge and embolize, resulting in stroke or other thromboembolic complications. Alternately, it can dissolve over time, aided by intrinsic fibrinolytic mechanisms. On other occasions, the thrombus may organize, a process characterized by ingrowth of smooth muscle cells, fibroblasts, and endothelium.