Dual antiplatelet therapy for acute coronary syndromes: How long to continue?





ABSTRACT
For patients with an acute coronary syndrome event, current guidelines recommend dual antiplatelet therapy for at least 12 months after drug-eluting stent placement. However, several clinical trials have assessed whether continuing dual antiplatelet therapy beyond 12 months is beneficial. We review the pros and cons of extending dual antiplatelet therapy.
KEY POINTS
- The outcomes of patients with acute coronary syndrome events have been improving as percutaneous coronary intervention and its accompanying medical therapy have evolved.
- Newer, more potent antiplatelet agents are preferred over clopidogrel when possible.
- Two earlier studies showed no advantage of extended dual antiplatelet therapy over the standard 12-month duration, but the recent Dual Antiplatelet Therapy trial did.
- The protection against ischemia afforded by dual antiplatelet therapy comes at the price of increased risk of bleeding.
Percutaneous coronary intervention for acute coronary syndromes has evolved, and so, hand in hand, has antiplatelet therapy. With the advent of clopidogrel and newer agents, several studies demonstrated the benefits of dual antiplatelet therapy in preventing major vascular ischemic complications. The findings culminated in a guideline recommendation for at least 12 months of dual antiplatelet therapy after placement of a drug-eluting stent, when feasible—a class I recommendation (treatment should be given), level of evidence B (limited populations evaluated).1,2 But extending dual antiplatelet therapy beyond 12 months had no strong favorable evidence until the recent Dual Antiplatelet Therapy (DAPT) study3 shed light on this topic.
Here, we review the evidence thus far on the optimal duration of dual antiplatelet therapy in the secondary prevention of coronary artery disease.
PLATELETS IN ACUTE CORONARY SYNDROMES AND STENT THROMBOSIS
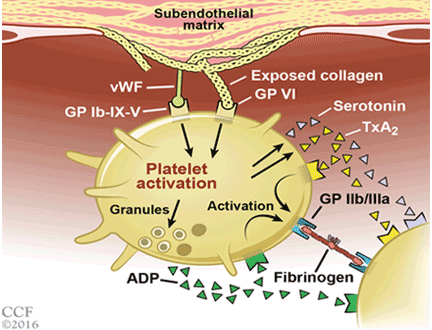
Acute coronary syndromes begin with fissuring or ulceration of a vulnerable atherosclerotic plaque, followed by thrombosis and occlusion, mediated by platelet adhesion, activation, and aggregation (Figure 1). Transient occlusion results in unstable angina or non-ST-elevation myocardial infarction, while total occlusion usually results in ST-elevation myocardial infarction.
Platelet aggregation is prominent among the mechanisms leading to stent thrombosis and vaso-occlusive ischemic complications after percutaneous coronary intervention. Thus, antiplatelet agents play a vital role in both primary and secondary prevention of cardiovascular events.4–6
Adhesion, activation, and aggregation
Adhesion. Disruption of the vascular endothelium as a result of vulnerable plaque fissuring or ulceration exposes subendothelial thrombogenic collagen and von Willebrand factor to blood. Collagen engages platelets through their glycoprotein (GP) Ia, IIa, and VI receptors, and von Willebrand factor binds platelets through the GP Ib-IX-V receptor.
Activation. Once platelets adhere to the subendothelium, they undergo a conformational change and become activated. Simultaneous release of various autocrine and paracrine mediators including adenosine diphosphate, serotonin, epinephrine, thromboxane, and various ligand-receptor interactions all contribute to the activation cascade. Adenosine diphosphate binds to the platelet receptor P2Y1, leading to an increase in intracellular calcium, and it binds to P2Y12, leading to a decrease in cyclic adenosine monophosphate, both of which cause GP IIb/IIIa receptor activation. Thromboxane A2 released by platelets by cyclo-oxygenase 1 binds to alpha or beta variant receptors and contributes to GP IIb/IIIa activation through elevation of intracellular calcium levels.
Aggregation and thrombosis. Exposure of tissue factor to plasma following plaque rupture activates the coagulation cascade via the extrinsic pathway, which generates thrombin, a powerful platelet activator that causes thrombus formation via fibrin. Thrombin binds to protease-activated receptors PAR-1 and PAR-4 on platelets, causing an increase in intracellular calcium and a decrease in cyclic adenosine monophosphate with subsequent GP IIb/IIIa activation. GP IIb/IIIa facilitates platelet aggregation by binding to fibrinogen and forming a stable platelet thrombus.
In the early stages of thrombus formation, platelets predominate (“white” thrombi); further organization with fibrin results in older “red” thrombi. The stages of thrombi vary in non-ST-elevation and ST-elevation myocardial infarction and are prognostic markers of death.4–8
PERCUTANEOUS INTERVENTION, RESTENOSIS, AND STENT THROMBOSIS
Percutaneous coronary intervention, the preferred means of revascularization for many patients, is performed emergently in patients with ST-elevation myocardial infarction, urgently in those with acute coronary syndromes without ST elevation, and electively in those with stable ischemic symptoms.
Percutaneous revascularization techniques have evolved from balloon angioplasty to bare-metal stents to drug-eluting stents, but each of these procedures has been associated with a periprocedural and postprocedural risk of thrombosis.

Balloon angioplasty was associated with vascular intimal injury, inciting elastic vascular recoil and smooth muscle cell proliferation leading to restenosis.
Bare-metal stents reduced the restenosis rate by eliminating vascular recoil, although restenosis still occurred within the stent because of neointimal proliferation of vascular smooth muscle cells. This was an important limitation, as both acute and subacute stent thrombosis were refractory to aggressive anticoagulation regimens that were associated with major bleeding complications and longer hospital length of stay. Stenting became mainstream practice only after the ISAR9 and STARS10 trials showed that dual antiplatelet therapy controlled stent thrombosis.
Drug-eluting stents coated with anti-proliferative and anti-inflammatory polymers markedly reduced in-stent restenosis rates by suppressing the initial vascular smooth-muscle proliferative response. However, they were still associated with late and very late stent thrombosis with incomplete endothelialization, even up to 40 months after implantation. Proposed mechanisms include incomplete stent apposition and inflammatory hypersensitivity reactions to the polymer coating. Incomplete stent apposition associated with low-velocity blood flow at the junction of the stent strut and vessel wall, together with delayed endothelialization, promotes platelet adhesion and aggregation, followed by thrombus formation.11
Second-generation drug-eluting stents have thinner struts and more biocompatible polymers and are thought to favor more complete re-endothelialization, reducing the rates of stent thrombosis.8,12,13
Predictors of early stent thrombosis
The Dutch Stent Thrombosis Registry and other studies looked at risk factors for stent thrombosis.14,15
Procedure-related factors included:
- Stent undersizing
- Residual uncovered dissections after angioplasty
- Longer stents
- Low flow after angioplasty (< 3 on the 0–3 Thrombolysis in Myocardial Infarction [TIMI] scale).
Lesion-related factors included:
- Intermediate coronary artery disease both proximal and distal to the culprit lesions
- Bifurcation lesions.
Patient-related factors included:
- Low left ventricular ejection fraction
- Diabetes mellitus
- Peripheral arterial disease Premature discontinuation of clopidogrel.
ANTIPLATELET AGENTS: MECHANISM OF ACTION
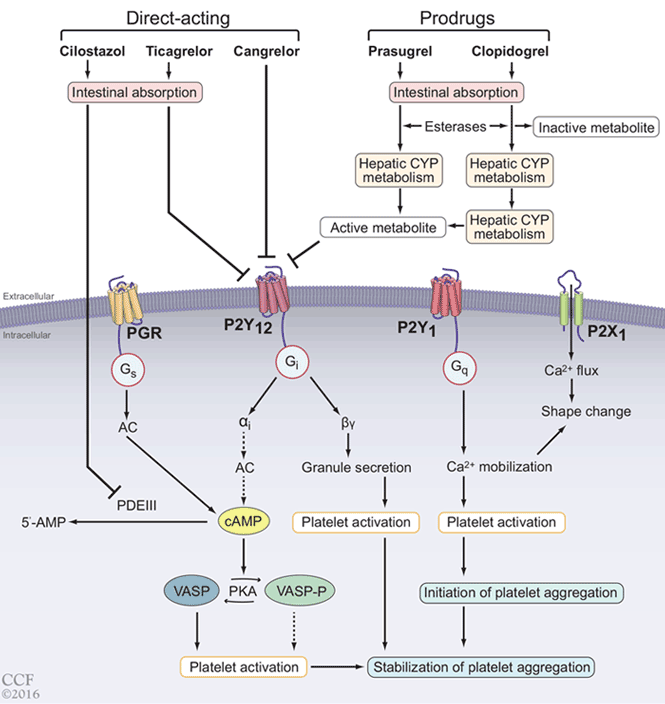
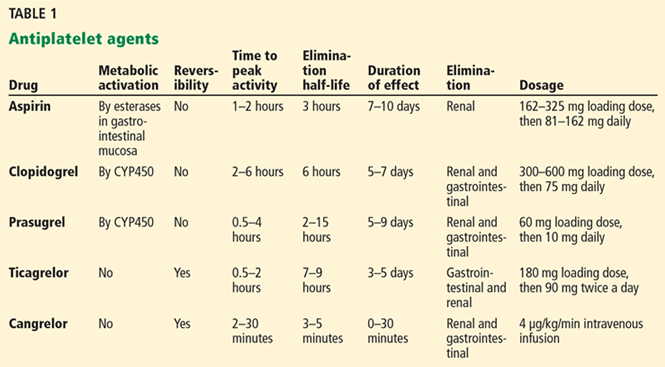
Various pathways play synergistic roles in platelet activation and aggregation and thrombus formation, and different antiplatelet agents inhibit these specific pathways, thus complementing each other and having additive effects (Figure 2, Table 1).5,16–21
Aspirin inhibits cyclo-oxygenase 1
Cyclo-oxygenase 1, found in platelets, endothelial cells, and other cells, catalyzes the conversion of arachidonic acid to thromboxane A2. Aspirin irreversibly inhibits cyclo-oxygenase 1 by acetylating its serine residue, preventing formation of thromboxane A2 and preventing platelet activation and aggregation.
P2Y12 ADP receptor antagonists
Clopidogrel and prasugrel are thienopyridine agents that irreversibly inhibit the P2Y12 receptor, thereby preventing binding of adenosine diphosphate and the subsequent platelet activation-aggregation cascade. They are both prodrugs and require conversion by cytochrome P450 enzymes to active metabolites. Prasugrel is 10 times more potent than clopidogrel due to more efficient formation of its active metabolite, and it achieves a comparable effect on platelet inhibition 30 minutes faster than the peak effect of clopidogrel at 6 hours. The overall peak inhibitory effect of prasugrel is twice that of clopidogrel.22
Ticagrelor, a cyclopentyl-triazolo-pyrimidine, directly and reversibly inhibits the P2Y12 ADP receptor. Unlike clopidogrel and prasugrel, it does not need to be converted to an active metabolite, and it noncompetitively inhibits P2Y12 at a site different from the adenosine diphosphate binding site.23 Like prasugrel, ticagrelor inhibits platelet function more rapidly and more completely than clopidogrel.
Cangrelor, an intravenously administered analogue of adenosine triphosphate, reversibly inhibits the P2Y12 receptor. It has undergone phase 3 trials but is not yet approved for clinical use.24
WHY DUAL ANTIPLATELET THERAPY?
Aspirin is good, clopidogrel is better
Aspirin has a well-validated role in both primary and secondary prevention of coronary and noncoronary atherosclerotic vascular disease.
The CAPRIE trial found clopidogrel monotherapy to be superior to aspirin monotherapy in patients with established atherosclerotic vascular disease.25
After stenting, short-term dual therapy is better than short-term warfarin
Thrombotic complications in the early postprocedural period were a major limitation of stenting, and existing anticoagulation regimens were ineffective in preventing them.26,27
The ISAR trial studied the benefit of combined antiplatelet vs anticoagulant therapy after stent placement. Patients randomized to receive combined aspirin plus ticlopidine (an early P2Y12 inhibitor) had significantly lower rates of primary cardiac, hemorrhagic, and vascular events at 30 days.9 Two other trials confirmed this finding.28,29
STARS10 also confirmed the benefit of aspirin and ticlopidine after stenting. Patients were randomly assigned to aspirin alone, aspirin plus warfarin, or aspirin plus ticlopidine after stent placement. The rate of stent thrombosis at 30 days was significantly lower in the dual antiplatelet group than in the other two groups. The dual antiplatelet group had a higher rate of bleeding than the aspirin-alone group, but the rate was similar to that of the aspirin-plus-warfarin group.
Long-term dual antiplatelet therapy is beneficial in several situations
ISAR and STARS were landmark trials that showed stent thrombosis could be reduced by dual antiplatelet therapy for a 30-day period. However, the long-term role of dual antiplatelet therapy was still unknown.
The CURE trial30–32 randomized patients presenting with acute coronary syndromes without ST elevation to receive clopidogrel plus aspirin or placebo plus aspirin for 3 to 12 months. The rate of the primary end point (cardiac death, nonfatal myocardial infarction, or stroke) was significantly lower in the clopidogrel-plus-aspirin group. A similar benefit of dual antiplatelet therapy was seen in the subgroup of patients who underwent percutaneous coronary intervention. Both pretreatment with clopidogrel plus aspirin for a median of 10 days prior to percutaneous intervention and continuing it for a mean of 9 months reduced major adverse cardiovascular events.
The CREDO trial20 found that the combination of clopidogrel and aspirin significantly reduced the incidence of death, myocardial infarction, or stroke at 1 year after percutaneous coronary intervention. A subgroup of patients in this trial who had a longer pretreatment interval with a loading clopidogrel dose showed a benefit at 28 days, which was not as evident with a shorter loading dose interval.
The CLARITY-TIMI 28 trial33,34 showed the advantage of adding clopidogrel to aspirin in patients receiving fibrinolytic therapy for ST-elevation myocardial infarction. Adding clopidogrel both improved the patency of the infarct-related artery and reduced ischemic complications. In patients who subsequently underwent percutaneous coronary intervention and stenting, clopidogrel pretreatment was associated with a significant decrease in ischemic complications before and after the procedure. There was no significant increase in bleeding complications in either group.
COMMIT/CCS 235 also showed the benefit of dual antiplatelet therapy in patients with ST-elevation myocardial infarction. Clopidogrel added to aspirin during the short-term in-hospital or postdischarge treatment period significantly reduced a composite end point of reinfarction, death, or stroke as well as death from any cause.
The CHARISMA trial36–38 aimed to determine if patients who were more stable (ie, no recent acute coronary syndrome event or percutaneous coronary intervention) would benefit. Overall, CHARISMA showed no benefit of adding clopidogrel to aspirin compared with aspirin alone in a broad population of patients with established vascular disease (secondary prevention) or risk factors for vascular disease (primary prevention).
But importantly, though no benefit was seen in the primary prevention group, the large subgroup of patients with established atherosclerotic vascular disease (12,153 of the 15,603 patients in the trial) did benefit from dual antiplatelet therapy.36,37 This subgroup showed an overall reduction in absolute risk of 1.5% (relative risk 0.88, P = .046) over a median follow-up of 27.6 months. This benefit was even more apparent in the 9,478 patients with prior myocardial infarction, stroke, or peripheral artery disease, for whom the relative risk reduction was 17.1% (P = .01) and the reduction in absolute risk 1.5%.38
These results are comparable to the 2% absolute risk reduction in the CURE trial for similar end points over 9 months. In both studies, there was no significant increase in the risk of major bleeding or intracranial bleeding in the clopidogrel-plus-aspirin groups, although minor bleeding was increased by dual antiplatelet therapy.
The rate of severe bleeding, which was the primary safety end point in CHARISMA, was not significantly different in the clopidogrel-plus-aspirin group compared with the placebo-plus-aspirin group (relative risk 1.25, 95% CI 0.97–1.61, P = .09).
Thus, although the CHARISMA findings were negative overall, the positive finding observed in the predominant subgroup of patients with established vascular disease can therefore be considered supportive of the results of the subsequent trials discussed below.