Anemia of chronic kidney disease: Treat it, but not too aggressively





ABSTRACTAnemia of renal disease is common and is associated with significant morbidity and death. It is mainly caused by a decrease in erythropoietin production in the kidneys and can be partially corrected with erythropoiesis-stimulating agents (ESAs). However, randomized controlled trials have shown that using ESAs to target normal hemoglobin levels can be harmful, and have called into question any benefits of ESA treatment other than avoidance of transfusions.
KEY POINTS
- Before treating with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies.
- Recognizing anemia in chronic kidney disease is important and often involves participation by the primary care physician, especially in early disease when chronic kidney disease may be mild.
- The only proven benefit of ESA therapy is avoidance of blood transfusions.
- ESAs should not be used to increase the hemoglobin concentration above 13 g/dL. In end-stage renal disease, the goal of therapy is to maintain levels at a target no higher than 11.5 g/dL. In nondialysis-dependent chronic kidney disease, the decision to prescribe ESA therapy should be individualized.
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
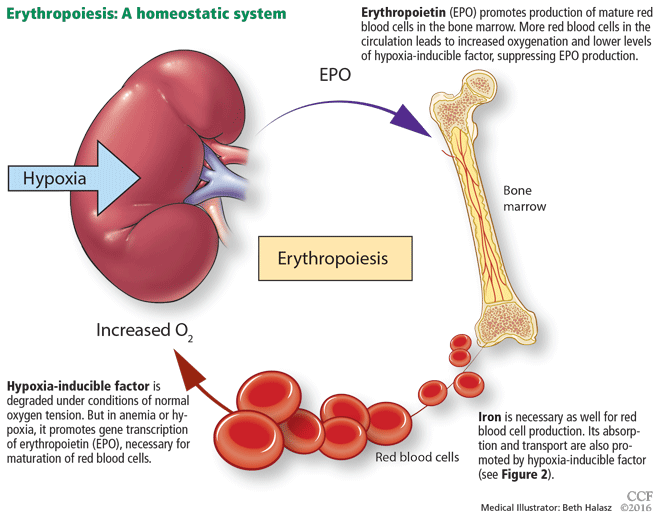
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
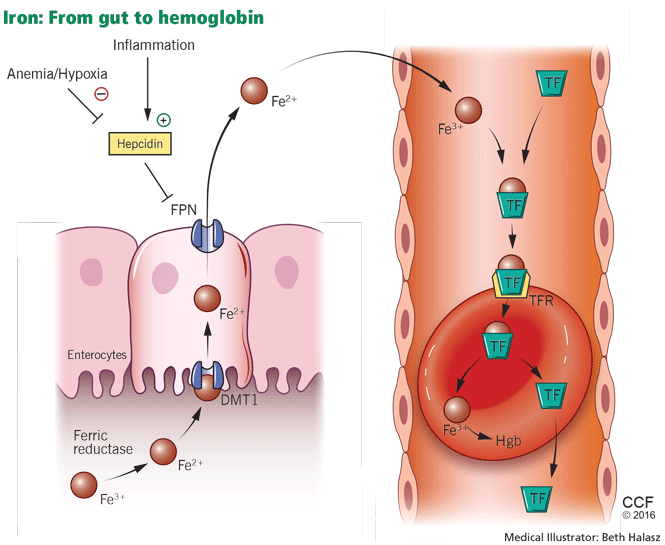
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
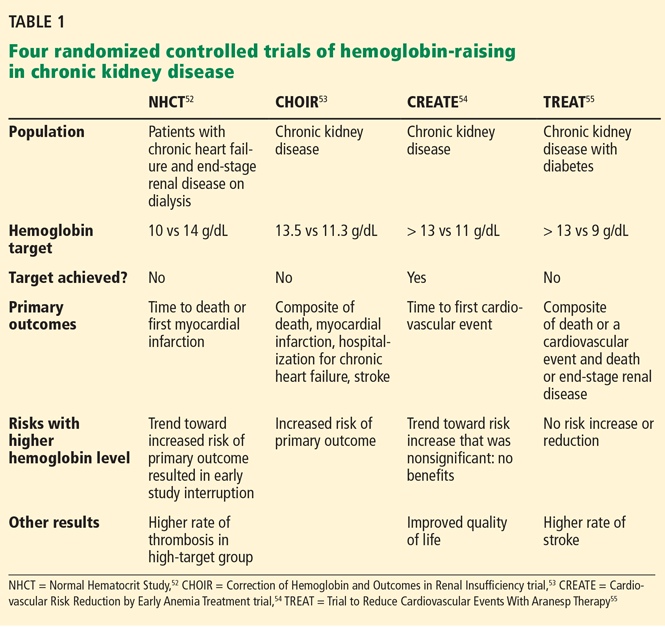
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.