Your patient has chronic leukemia: Now what?





ABSTRACTAlthough still in their infancy, biologic therapies for hematologic cancers are making rapid strides, diminishing the role of chemotherapy and offering long-term remission. More patients are surviving cancer and therefore are increasingly being seen by primary care physicians, who must be aware of complications of standard and newer treatments and how to manage them.
KEY POINTS
- Chronic myelogenous leukemia (CML) can now be functionally cured with tyrosine kinase inhibitors, which interfere with the product of the oncogene causing the disease.
- Patients diagnosed with CML should begin therapy immediately even if they have no symptoms.
- Tyrosine kinase inhibitors have side effects that increase cardiovascular risk.
- Chronic lymphocytic leukemia (CLL) is an immunologic disease involving clonal proliferation of B cells. Chemotherapy for CLL should begin only when symptoms or indicators of impaired marrow function reach a certain threshold.
- New treatments for CLL increase the risk of atrial fibrillation and autoimmunity.
- Experimental B-cell–targeted therapies have demonstrated encouraging results even when chemotherapy fails in CLL and other B-cell cancers.
The advent of targeted therapies has dramatically changed the management of chronic leukemia. Chemotherapy—highly toxic, nonspecific drugs that can be dangerous to patients and providers and result in only modest success—is gradually being replaced by biologic targeting of malignancy. Scientists are rapidly identifying extracellular and intracellular targets on tumor cells and are developing and testing promising new therapies aimed at these targets. Survival of cancer patients has become so common that clinicians outside the specialties of hematology and oncology are now caring for them.
This article describes new biologic therapies for chronic myelogenous leukemia (CML) and chronic lymphocytic leukemia (CLL), along with the diagnosis of these diseases and management of survivors in the primary care setting.
CHRONIC MYELOGENOUS LEUKEMIA
A seemingly healthy person needs laboratory blood work, perhaps for an insurance physical examination or for a preoperative workup. Or a patient comes to the emergency department with a sore throat and routine blood tests are ordered. Their laboratory values:
- White blood cell count 250 × 109/L (reference range 3–11)
- Neutrophils 70% (40%–70%)
- Blasts 1% (0)
- Metacytes and myelocytes 5% (0)
- Bands 5% (0)
- Lymphocytes 10% (22%–40%)
- Monocytes 5% (0–7%)
- Basophils 3% (0–1%)
- Eosinophils 1% (0–4%)
- Hemoglobin 12.1 g/dL (11.5–15.5 in women, 13.0–17.0 in men)
- Platelet count 525 × 109/L (150–400).
Leukocytosis and a ‘left shift’
Although this scenario often raises concern for acute leukemia, a careful look shows evidence of a chronic myeloproliferative disorder instead. Specifically, this patient’s laboratory values show a “left shift”—an increase in immature neutrophils, ie, blasts, myelocytes, and bands.
This picture is characteristic of CML, an uncommon leukemia with about 4,500 new cases annually in the United States. Patients can present at any age, but the disease occurs more often in older people, with a median age of 66.1
The presentation is usually subtle: about half of cases are detected by routine laboratory testing, which typically reveals a left-shifted leukocytosis with basophilia and a few blasts. Mild anemia is common. The platelet count is elevated in 30% to 50% of patients at diagnosis. Bone marrow aspirate shows significant myeloid hyperplasia without dysplasia, and sometimes shows mild fibrosis.
Philadelphia chromosome is diagnostic
A definitive diagnosis is made by demonstration of an abnormally short chromosome 22. Described in 1960 by Peter Nowell of the University of Pennsylvania and David Hugerford of the Institute for Cancer Research,2 this abnormality, called the Philadelphia chromosome, was the first specific genetic abnormality associated with a human cancer. Later, researchers used banding techniques to find that the Philadelphia chromosome results from a reciprocal translocation of genetic material between the BCR gene on chromosome 22 and the ABL1 gene on chromosome 9, t(9:22).3,4 The resulting chimeric gene, called BCR-ABL, codes for an oncogenic protein, a tyrosine kinase with constitutive activity.
The Philadelphia chromosome is present in 95% of patients with CML and can be found in all myeloid cell lineages, including erythrocytes, granulocytes, monocytes, and megakaryocytes as well as some cells of lymphocytic lineage, indicating that malignant transformation to CML takes place at the stem cell level.
The mutation causes several problems: the abnormal tyrosine kinase increases cell proliferation, inhibits apoptosis, and alters adhesion molecules in the stroma of the bone marrow, allowing immature cells to leak into the bloodstream. Most important, the mutation increases genomic instability so that additional mutations are likelier to occur over time, making it inevitable that, without treatment, the disease will progress to a fatal blast crisis within an average of 5 years of diagnosis.
CML has three clinical phases
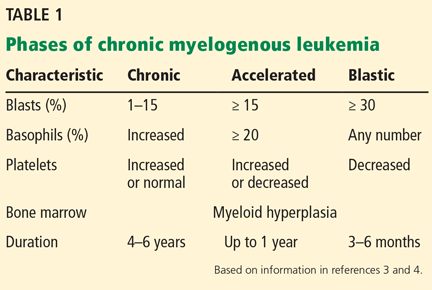
Untreated, CML progresses through three distinct phases: chronic, accelerated, and blast crisis, defined by abnormalities in the blood smear and bone marrow (Table 1).5,6 Most patients (85%) are diagnosed during the chronic phase. The accelerated and blastic phases resemble acute leukemia.
Chronic phase management
Therapies over the years have included arsenic (Fowler solution), splenic radiotherapy, busulfan, hydroxyurea, cytarabine, and interferon. All had some palliative success, but usually did not suppress leukemic progression.7
In contrast, patients undergoing allogeneic bone marrow transplant had a 5-year survival rate of 60% to 80% during the chronic phase of CML, 40% to 60% during the accelerated phase, and 10% to 20% during a blast crisis.8 Long-term survival confirmed the ability of transplant to cure CML, and bone marrow transplant with matched donors was the standard of care for younger patients until the end of the 20th century.
Tyrosine kinase inhibition
A new paradigm in treatment began with the development of imatinib, a tyrosine kinase inhibitor that directly interferes with the product of the chimeric BCR-ABL gene.9
Patients treated with imatinib during the chronic phase of CML have survival rates similar to those of people without the disease, and they usually do not progress to the accelerated and blast phases. As a result of this success, the number of transplants for CML has fallen precipitously.
Other tyrosine kinase inhibitors (dasatinib, nilotinib) that have since been developed have shown even better results in achieving remission and preventing progression. Improved survival is more difficult to demonstrate because the control groups in studies receive imatinib and have 10-year survival rates of about 90%.10–12
With the tyrosine kinase inhibitors, CML can be regarded as functionally cured.13 Patients take these drugs for life and usually experience a relapse if they stop. Patients with CML are now more likely to die of a comorbidity than of CML.
Choose therapy by tolerability
Which tyrosine kinase inhibitor to use depends more on the side-effect profile of the drug than on its efficacy. Nilotinib should be avoided in patients with vascular disease, and dasatinib avoided in patients with pulmonary disease. Each drug may be associated with some degree of nausea, diarrhea, cramps, rash, and edema.10–12
CML is not an immunosuppressive disease, nor are the drugs used to treat it. Patients with CML have an intact immune system. Therefore, precautions taken for patients with acute leukemia or lymphoid malignancy are not required for patients with CML.
Managing survivors
Since imatinib was introduced in 2000, the US Food and Drug Administration (FDA) has approved approximately 20 tyrosine kinase inhibitors for various cancers. These drugs are improving survival rates so well that patients with cancer are increasingly being seen by their primary care doctors for their medical problems.
Some problems have emerged that are consequences of this successful therapy. Angiogenesis inhibitors such as bevacizumab affect vascular endothelial growth factors, which injure endothelial cells. These effects may result in high blood pressure and arterial occlusive disease. Algorithms have been proposed for managing cardiovascular complications for patients taking tyrosine kinase inhibitors.14 Further, cardiovascular risk factors such as hyperlipidemia, diabetes, and obesity must be aggressively managed in patients taking tyrosine kinase inhibitors.
Vascular effects, rashes, and drug interactions may best be managed by primary care physicians, cardiologists, and nephrologists, who deal with such problems regularly.
CHRONIC LYMPHOCYTIC LEUKEMIA
A patient undergoes routine laboratory blood work in the emergency department or clinic, with these results:
- White blood cell count 250 × 109/L
- Neutrophils 1%
- Lymphocytes 99%
- Hemoglobin 12.1 g/dL
- Platelet count 160 × 109/L.
Like patients with CML, those with CLL usually present with no symptoms. The complete blood cell count reveals numerous white blood cells and lymphocytosis. Patients may have painless lymphadenopathy, anemia, and thrombocytopenia, but they do not typically have fever, sweats, or weight loss.
The disease is characterized by clonal proliferation and accumulation of mature-appearing neoplastic B lymphocytes in the blood, bone marrow, lymph nodes, and spleen. The peripheral blood smear shows “smudge cells,” indicating fragile lymphocytes.
The median age at diagnosis is about 70, with fewer than 15% of newly diagnosed patients under age 50.
CLL is the most common leukemia in the Western world, accounting for about 30% of cases of leukemia in adults. It is rare in Asians, probably because of genetic differences.
Monoclonal B-cell lymphocytosis precedes CLL
Monoclonal B-cell lymphocytosis is related to CLL and always precedes it. It is a common condition, detectable in up to 5% of older adults. The differential count shows a less severe lymphocytosis than in CLL.
Because monoclonal B-cell lymphocytosis does not always convert to leukemia, it is important for insurance coverage purposes not to diagnose it as a leukemia. Treatment-free survival of patients diagnosed with monoclonal B-cell lymphocytosis is 87% at 5 years.15,16