A tale of two sisters with liver disease





CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
,4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
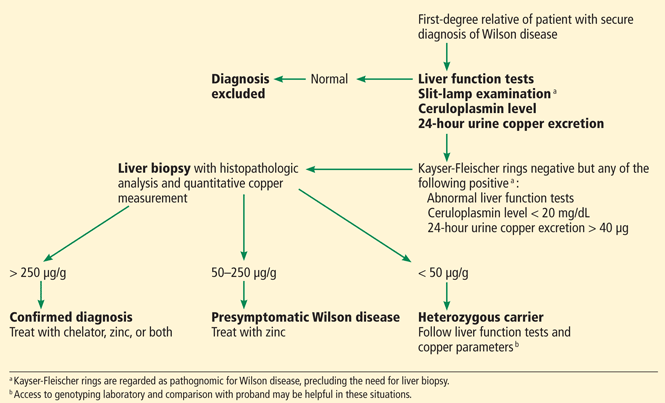
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
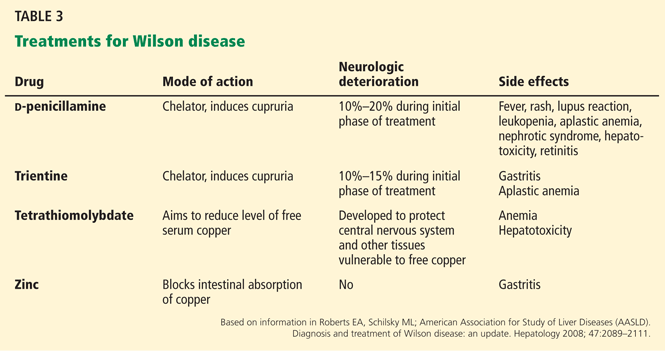
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
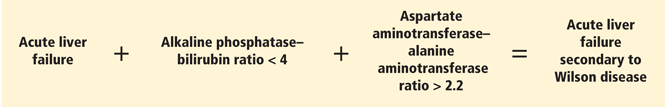
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.