Autoantibody-mediated encephalitis: Not just paraneoplastic, not just limbic, and not untreatable





ABSTRACTAutoantibody-mediated encephalitis is a heterogeneous group of recently identified disorders, all caused by autoimmunity directed against components of the central nervous system. Despite severe and even prolonged neurologic deficits, dramatic improvements may occur with aggressive treatment.
KEY POINTS
- Autoantibody-mediated encephalitis accounts for a portion of cases of unexplained status epilepticus, encephalitis, and acute-onset psychiatric symptoms.
- Magnetic resonance imaging and cerebrospinal fluid analysis may be normal early in the disease course.
- Patients can express more than one autoantibody and present with more than one neuronal syndrome.
- Syndromes in which antibodies attack antigens on the surface of neurons are more likely to respond to immunotherapy than those involving intracellular antigens.
- Anti-N-methyl-d-aspartate receptor encephalitis typically presents with psychosis, seizures, and movement disorders in young women and is often associated with an ovarian teratoma.
- Limbic encephalitis, mediated by antibody to the voltage-gated potassium channel complex, is typically nonneoplastic and responds well to immunotherapy.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
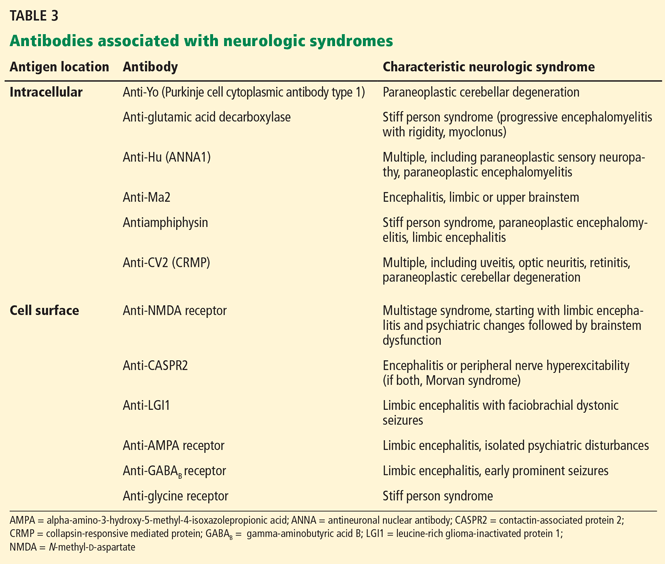
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.