Trastuzumab-dkst approval adds to the biosimilar cancer drug market





Citation JCSO 2018;16(2):e63-e65
©2018 Frontline Medical Communications
doi https://doi.org/10.12788/jcso.0398
Related articles
Biosimilars: same ol’ – but with a suffix, and cheaper
Submit a paper here
The human epidermal growth factor receptor-2 (HER2)-targeting monoclonal antibody trastuzumab-dkst, was approved by the US Food and Drug Administration in 2017 for the treatment of patients with HER2-positive breast or metastatic gastric or gastroesophageal junction adenocarcinoma.1 Trastuzumab-dkst, marketed as Ogviri by Mylan NV and Biocon Ltd, is a copy, known as a biosimilar, of Genentech’s trastuzumab (Herceptin), which has been approved in the US since 1998. Genentech’s patent on trastuzumab expires in 2018
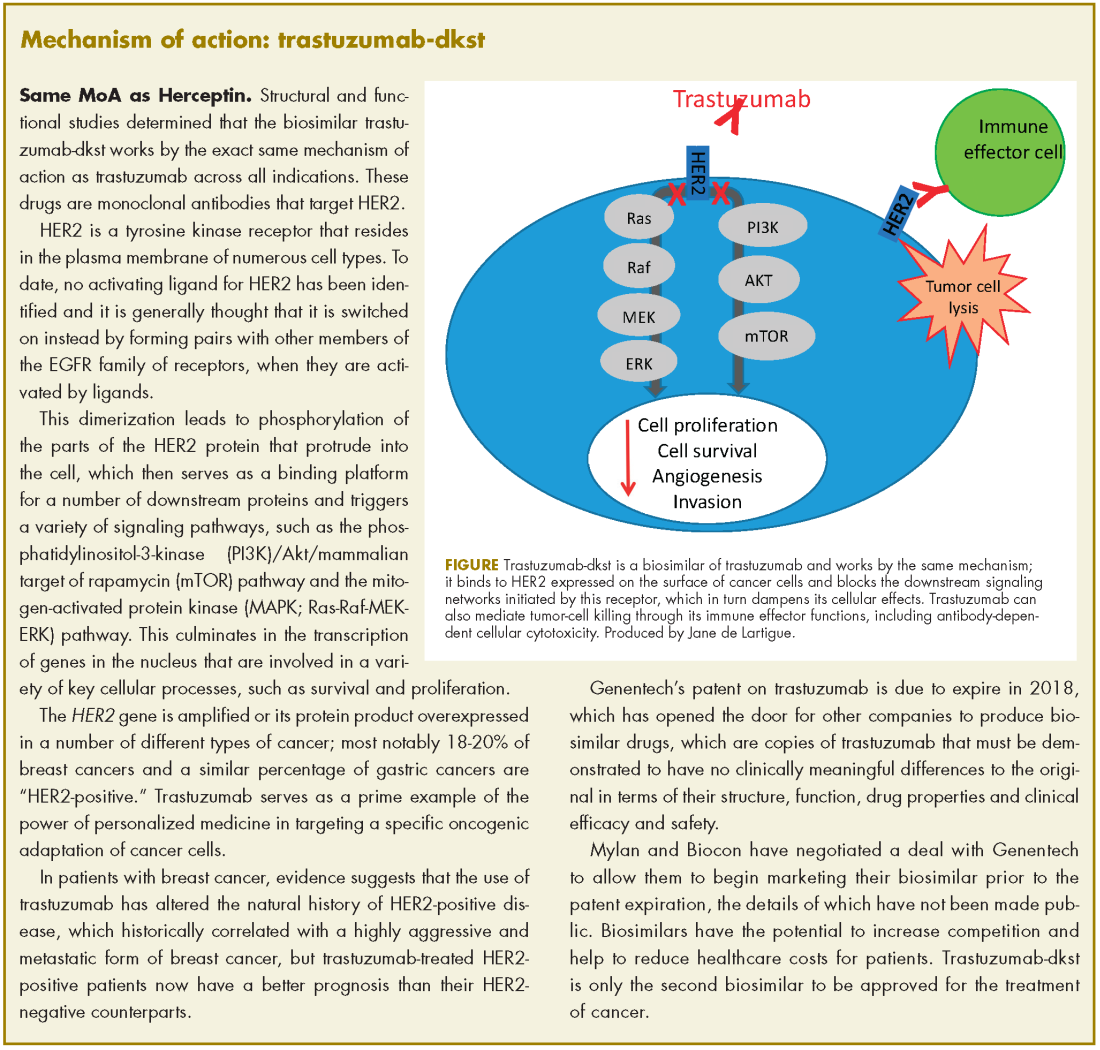
Approval was based on a comparison of the 2 drugs, which demonstrated that there were no clinically meaningful differences between the biosimilar and the reference product (trastuzumab) in terms of structure and function, pharmacokinetics (PKs), pharmacodynamics, and clinical efficacy and safety.
,In structural and functional studies, trastuzumab-dkst was shown to have an identical amino acid sequence and a highly similar 3-dimensional structure, as well as equivalency in an inhibition of proliferation assay, a HER2-binding assay, and an antibody-dependent cellular cytotoxicity assay, compared with trastuzumab.
Two nonclinical animal studies were performed in cynomolgus monkeys; a single-dose comparative PK study and a 4-week, repeat-dose toxicity study. That was further supported by data from a single-dose, randomized, double-blind, comparative 3-way PK study (MYL-HER-1002) in which 120 healthy men were given an 8 mg/kg infusion of trastuzumab-dkst, US-approved trastuzumab, or European Union (EU)-approved trastuzumab.
The key clinical study was the phase 3 HERiTAge trial, a 2-part, multicenter, double-blind, randomized, parallel group trial that was performed in patients with HER2-positive metastatic breast cancer who had not been previously treated with either chemotherapy or trastuzumab in the metastatic setting.2
Eligible patients included males or females with measurably HER2-positive disease (as defined by HER2 overexpression determined by immunohistochemistry performed by a central laboratory), no exposure to chemotherapy or trastuzumab in the metastatic setting, an Eastern Cooperative Oncology Group Performance Status of 0 or 2, left ventricular ejection fraction (LVEF) within institutional range of normal, and who had completed adjuvant trastuzumab therapy at least 1 year before.
Patients with central nervous system metastases had to have stable disease after treatment, and hormonal agents were required to be discontinued before the start of the study. Patients with a history of unstable angina, heart failure, myocardial infarction less than 1 year from randomization, other clinically significant cardiac disease, grade 2 or higher peripheral neuropathy, a history of any other cancer within 4 years before screening, or any significant medical illness that increased treatment risk or impeded evaluation, were excluded from the study.
Patients were randomly assigned 1:1 to receive trastuzumab-dkst or trastuzumab, both in combination with paclitaxel or docetaxel, at a loading dose of 8 mg/kg, followed by a maintenance dose of 6 mg/kg, every 3 weeks for a minimum of 7 cycles in part 1 of the study. Patients who had stable disease or better were enrolled in part 2 and continued treatment until disease progression or unacceptable toxicity.
The primary endpoint was overall response rate (ORR) and, after 24 weeks, the ORR was 69.6% in the trastuzumab-dkst arm, compared with 64% in the trastuzumab arm, with a ratio of ORR of 1.09. Progression-free survival was also nearly identical in the 2 groups and median overall survival had not been reached in either arm.
The safety of the biosimilar and reference product were also highly similar. Serious adverse events occurred in 39.3%, compared with 37% of patients, respectively, with neutropenia the most frequently reported in both arms. Overall, treatment-emergent AEs occurred in 96.8%, compared with 94.7% of patients, respectively, with the majority of events mild or moderate in severity in both groups. This study also confirmed the low immunogenicity of the 2 drug products.
The prescribing information details the recommended doses of trastuzumab-dkst for each approved indication and warnings and precautions for cardiomyopathy, infusion reactions, pulmonary toxicity, exacerbation of chemotherapy-induced neutropenia and embryofetal toxicity.3
Patients should undergo thorough cardiac assessments, including baseline LVEF measurement immediately before starting therapy, every 3 months during therapy, and upon completion of therapy. Patients who complete adjuvant therapy should have cardiac assessments every 6 months for at least 2 years. Treatment should be withheld for ≥16% absolute decrease in LVEF from pre-treatment values or an LVEF value below institutional limits of normal and ≥10% absolute decrease in LVEF from pre-treatment values. When treatment is withheld for significant LVEF cardiac dysfunction, patients should undergo cardiac assessment at 4-week intervals.
To combat infusion reactions, infusion should be interrupted in all patients experiencing dyspnea or clinically significant hypotension and medical therapy administered. Patients should be evaluated and monitored carefully until signs and symptoms resolve and permanent discontinuation considered in patients with severe reactions. Patients should be warned of the potential for fetal harm with trastuzumab-dkst and of the need for effective contraceptive use during and for 6 months after treatment