Atraumatic splenic rupture as an initial presentation of chronic myelogenous leukemia





Accepted for publication April 4, 2016. Correspondence Erik Frost, DO; erikjfrost@gmail.com. Disclosures The authors report no disclosures or conflicts of interest.
JCSO 2017;15(2):113-115. ©2017 Frontline Medical Communications. doi: https://doi.org/10.12788/jcso.0263.
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm associated with the fusion of the BCR gene located on chromosome 22 and the ABL1 gene on chromosome 9. The fusion results in a reciprocal translocation between chromosomes 9 and 22, leading to the formation of the Philadelphia (Ph) chromosome found in 90%-95% of patients with CML. The incidence of CML is 1.5 per 100,000 people per year, with a male predominance and an average age at diagnosis of 64.1
About 85%-90% of newly diagnosed patients present in the chronic phase and therefore many of them are asymptomatic at the time of diagnosis. If symptoms are present, they often include fatigue, malaise, unintentional weight loss, early satiety, or left upper quadrant pain. Progression of the disease is associated with worsening symptoms such as unexplained fever, significant weight loss, bone or joint pain, bleeding, thrombosis, and infections suggestive of transformation to the accelerated phase or blast crisis. Physical exam findings most commonly include splenomegaly and occasionally mild hepatomegaly.
Atraumatic splenic rupture is a rare complication of this hematologic malignancy, and there are almost no reported cases of CML as the underlying cause.2-4 Here we present the case of a man with sudden-onset generalized abdominal pain and leukocytosis. A computed-tomography scan showed splenic rupture, and the patient was taken for emergency splenectomy. The patient was subsequently positive for t(9,22)(q34;q11.2).
,Case presentation and summary
A 59-year-old white man with a history of hypertension and kidney stones presented to a community emergency department with a chief complaint of abdominal pain. About 30 minutes before his arrival, the patient had woken up from sleep with generalized, nonradiating, abdominal pain, which he described as “like my previous kidney stones.” He also reported worsening dyspnea, nausea without vomiting, and lightheadedness without loss of consciousness. The remainder of the review of systems was negative. A physical exam revealed that he was in moderate distress with clear lung fields and had tachycardia without murmur, no CVA tenderness, and a diffusely tender abdomen.
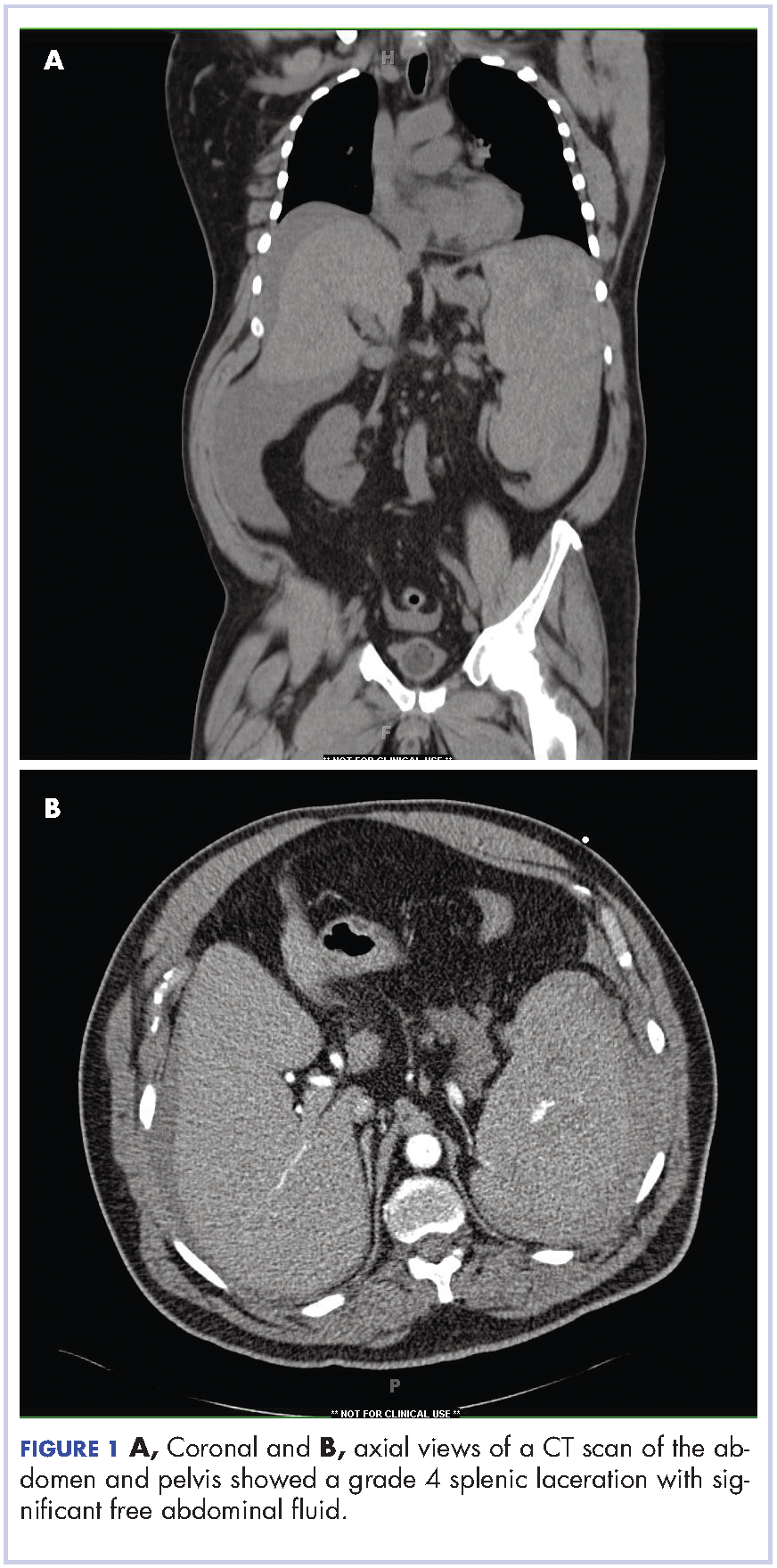
Complete blood count with differential showed leukocytosis (109.1 x 103/uL), normocytic anemia (8.1 g/dL), thrombocytopenia (100,000 cells/uL), neutrophils (71.06 cells/uL), bands (27.13 cells/uL), and monocytes (11.63 cells/uL). A CT scan of the abdomen and pelvis showed a grade 4 splenic laceration with significant free abdominal fluid (Figure 1).
The patient was taken to the operating room where he underwent a splenectomy which was complicated by partial gastrectomy and partial omentectomy. He remained intubated on mechanical ventilation in the intensive care for 7 days. His progress was complicated by profound hypotension that required significant fluid administration and ultimately multiple pressors for blood pressure support. Hypotensive shock was beginning to improve on day 3 and was completely resolved by day 5. The patient underwent continuous positive airway pressure (CPAP) trials on day 6 and was successfully extubated on day 7.
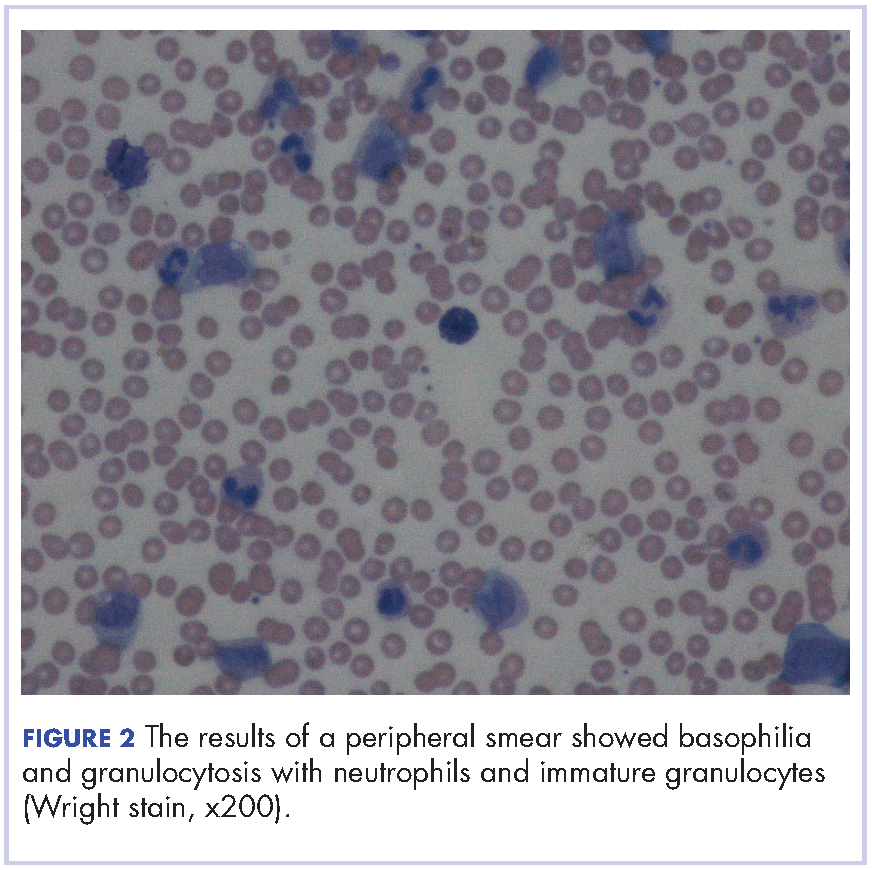
After extubation a more thorough history could be obtained from the patient. He denied any history of weight loss, night sweats, or fatigue. Patient denied any known family history of hematologic malignancies. His peripheral smear showed basophilia and granulocytosis with neutrophils and immature granulocytes (Figure 2). The patient was evaluated by the hematology service and was started on allopurinol and hydroxyurea for presumed hematologic malignancy. He was given the meningococcus and streptococcus pneumoniae vaccine and was discharged home in stable condition on day eleven. Patient was subsequently positive for t(9,22)(q34;q11.2) and was started on imatinib. He has continued to follow in the clinic and is currently in remission.
Discussion
CML has a triphasic clinical course and treatment is based on the specific disease phase. The 3 phases of the disease include the chronic (more indolent) phase, accelerated (more aggressive) phase, and blast crisis. If the disease is left untreated, it will inevitably transition from a chronic to an accelerated phase and finally to blast crisis within a median time of 4 years.
The chronic phase is the most common, representing 85% of diagnoses. Patients can be asymptomatic and many in this phase will be diagnosed by routine lab testing.5 According to the World Health Organization, the accelerated phase is defined as CML patients with one of the following: 10%-19% blasts, basophils ≥20%, platelets <100,000/microL or >1,000,000/microL, unresponsive to therapy, splenomegaly unresponsive to therapy, an increasing white cell count unresponsive to therapy, or cytogenetic evolution.6 Blast crisis is the most aggressive phase and is usually defined by ≥20% blasts, large foci or clusters of blasts on the bone marrow biopsy, or the presence of extramedullary blastic infiltrates.7,8
The diagnosis of CML should be suspected in the presence of distinct lab abnormalities in the peripheral blood. These include elevated white blood cell counts with a median count of 100,000 cells/microL, elevated platelet counts, and a mild normocytic normochromic anemia. Platelet counts of 600,000 or greater have been seen in 15%-30% of patients at the time of diagnosis. The white count differential can show a variety of cells but there will be a notably greater percentage of myelocytes than metamyelocytes. Bone marrow biopsy will reveal increased cellularity, normal to slightly elevated percentage of blasts, and reticulin fibrosis. The diagnosis should be confirmed by the presence of the Philadelphia chromosome either by cytogenetics, fluorescence in situ hybridization, or reverse-transcription polymerase chain reaction (RT-PCR). The Philadelphia chromosome is found in 90%-95% of patients with CML. Most of the remaining patients will have other translocations, but a small minority will have no detectable genetic abnormalities and those patients are known as Ph-negative.9
Treatment options for CML include potential cure with allogeneic hematopoietic stem-cell transplant (HSCT) or disease control using tyrosine kinase inhibitors (TKIs). TKIs are the initial treatment of choice for newly diagnosed patients and are able to produce long-term remission in most patients. The drugs in this category include imatinib, dasatinib, and nilotinib. They work by inhibiting the Bcr-Abl tyrosine kinase, thereby blocking proliferation and inducing apoptosis in Bcr-Abl-positive cells. The majority of patients with chronic-phase CML will have an excellent response to initial treatment with a TKI. It is critical to follow these patients on a regular basis and monitor their disease status. Although the gold standard for assessing cytogenetic response is cytogenetic analysis of a bone marrow biopsy, more sensitive methods such as quantitative PCR using peripheral blood are now available, thereby minimizing the need for bone marrow biopsy. Patients in the accelerated phase are more difficult to manage because they are resistant to most forms of treatment and have short-lived responses to TKI therapy. These patients should strongly be considered for transplantation. Patients in blast crisis have aggressive disease that is more complex and requires more extensive testing. These patients should ideally be treated at tertiary care centers and treatment often involves chemotherapy in addition to TKI therapy usually followed by HSCT.
Atraumatic splenic rupture (ASR) presents similarly to traumatic splenic rupture with typical symptoms being acute onset of upper abdominal, left chest wall, or left shoulder pain (Kehr’s sign) but without a known history of trauma. Quick recognition and surgical intervention represent the best means of definitive care.10 Renzulli and colleagues conducted a literature review for all ASR cases from 1980-2008, examining 632 publications representing 845 cases. They examined the cases using logistic regression analysis to better define the clinicopathology behind ASR. The reported causes of ASR are neoplastic processes (30.3%), infectious (27.3%), inflammatory noninfectious (20.0%), drug- and treatment-related (9.2%), mechanical (6.8%), and normal spleen (6.4%). Treatment included total splenectomy in 84.1% of cases, organ-preserving surgery in 1.2%, and conservative measures in 14.7%. They reported an ASR-related mortality of 12.2%, with being older than 40 and neoplastic disorders associated with increased mortality – although male sex and splenomegaly have also been reported.11-13 Thomas and colleagues have reported on 48 cases of ASR related to hematologic malignancy showing acute myeloid leukemia being the most common cause (21%), followed by acute lymphoblastic leukemia (19%).2
Hematologic malignancies commonly cause splenic engorgement and pain although splenic rupture is an extremely rare event. Recent literature review has shown fewer than a thousand reported cases since 1980.4 There far fewer reported cases of ASR being related to CML, with most being reported as a complication.3,14 Based on our review, we could identify only a handful cases of CML with ASR being the initial symptom. These include a patient with Ph-negative CML and ASR following blast crisis, a patient with Phil-negative BCR-ABL-positive essential thrombocythemia, several cases in which the patient ultimately died, and 1 in which the patient survived into remission.4,14-16 Our case is different because the patient was ultimately positive for t(9,22)(q34;q11.2) and although he experienced multiple complications, he is currently functioning at his baseline and in remission. We hope this case will remind others that CML should be considered in the differential diagnosis of patients ASR.