Progressive, pruritic eruption of firm, skin-colored papules






Scleromyxedema, or generalized lichen myxedematosus, is a primary cutaneous mucinosis with unknown pathogenesis characterized by generalized firm, skin-colored papules and is commonly associated with an underlying monoclonal gammopathy (usually Ig-gamma paraproteinemia).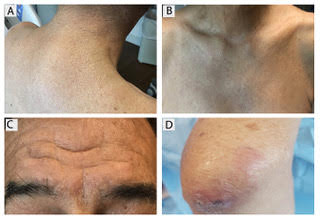
Scleromyxedema may have associated internal involvement, including neurologic, gastrointestinal, pulmonary, renal, cardiovascular, ophthalmological, or musculoskeletal. Histopathology demonstrates mucin in the dermis seen with Alcian blue staining, proliferation of fibroblasts, and increased collagen deposition.
The condition is chronic and progressive. Intravenous immunoglobulin is considered first-line treatment. Thalidomide and corticosteroids have been reported to also be efficacious.
It is associated with hematologic disorders, including IgA monoclonal gammopathy, as well as myeloproliferative disorders, leukemia, infections, and inflammatory bowel disease. Although its pathophysiology is not well understood, vascular immune complex deposition, repetitive inflammation, and subsequent fibrosis may play a role. On histology, there is leukocytoclastic vasculitis with polymorphonuclear cell infiltrate and fibrin deposition in the superficial and mid-dermis and onion-skin fibrosis.
EED often self-resolves within 5-10 years, although it can become chronic and recurrent. Dapsone, niacinamide, antimalarials, NSAIDs, tetracyclines, corticosteroids, colchicine, and plasmapheresis are reported treatments. This patient’s EED was recalcitrant to prednisone and responded to colchicine.
Scleromyxedema and EED are both rare, distinct cutaneous entities associated with different underlying paraproteinemias and to the best of our knowledge, have not been previously reported to coexist in a single patient.

This case and the photos were submitted by Rachel Fayne, BA; Yumeng Li, MD, MS; Fabrizio Galimberti, MD, PhD; and Brian Morrison, MD, of the department of dermatology and cutaneous surgery at the University of Miami.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.