Allergic Contact Dermatitis With Sparing of Exposed Psoriasis Plaques





Practice Points
- Patients with plaque-type psoriasis who experience allergic contact dermatitis (ACD) may present with sparing of exposed psoriatic plaques.
- The divergent immunologic milieus present in ACD and psoriasis likely underly the decreased incidence of ACD in patients with psoriasis.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
,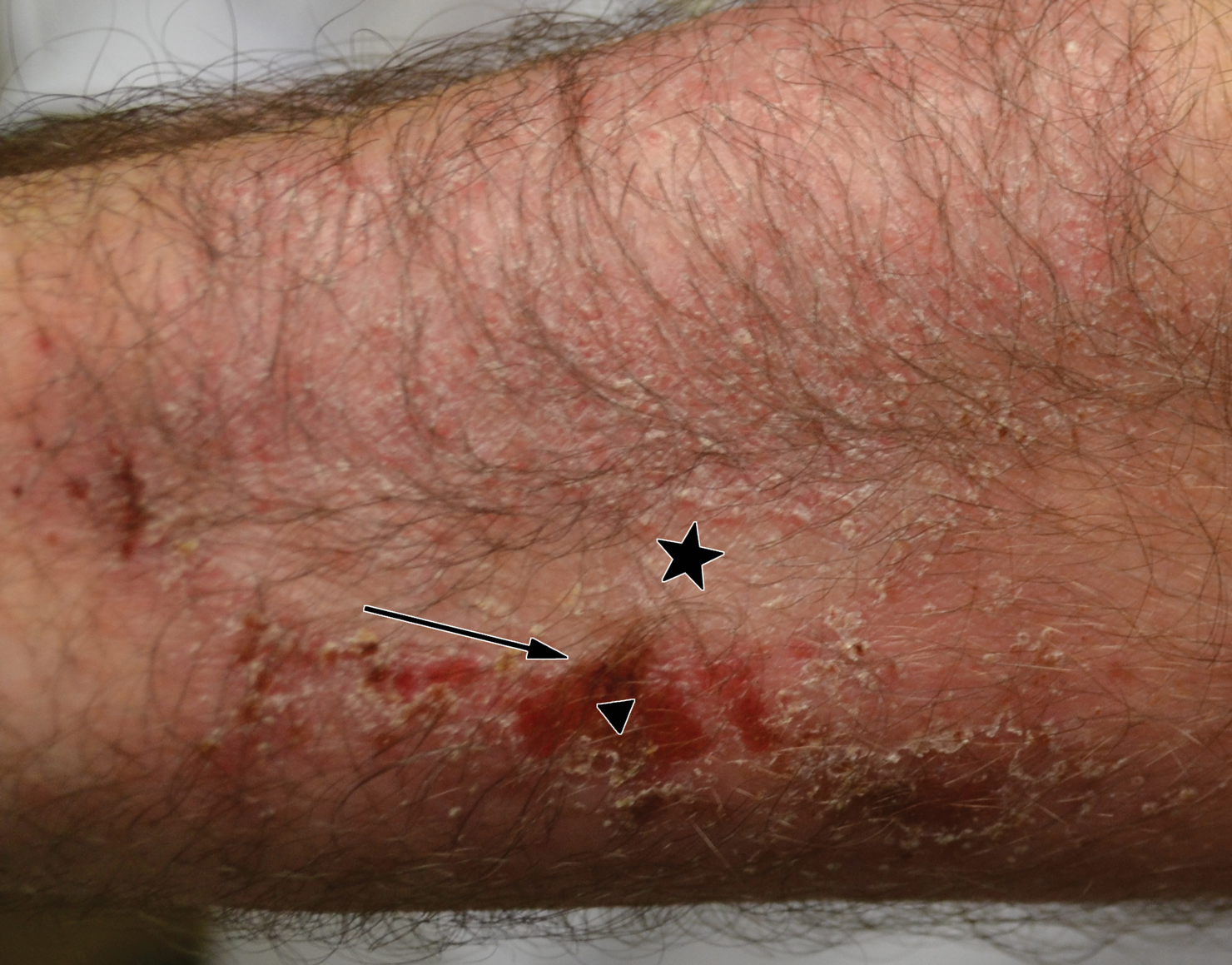
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.