Smooth Symmetric Plaques on the Face, Trunk, and Extremities





THE DIAGNOSIS: Lepromatous Leprosy
Histopathology showed collections of epithelioid to sarcoidal granulomas throughout the dermis and clustered around nerve bundles with a grenz zone at the dermoepidermal junction. Fite stain was positive for acid-fast bacteria, which were confirmed to be Mycobacterium leprae by by the National Hansen’s Disease program. Based on these findings, a diagnosis of lepromatous leprosy (LL) was made. The patient was treated by the infectious disease department with multidrug therapy that included monthly rifampin, moxifloxacin, and minocycline; weekly methotrexate with daily folic acid; and an extended prednisone taper with prophylactic cholecalciferol.
Lepromatous leprosy is characterized by high antibody titers to the acid-fast, gram-positive bacillus Mycobacterium leprae as well as a high bacillary load.1 Patients typically present with muscle weakness, anesthetic skin patches, and claw hands. Patients also may present with foot drop, ulcerations of the hands and feet, autonomic dysfunction with anhidrosis or impaired sweating, and localized alopecia.2 Over months to years, LL may progress to extensive sensory loss and indurated lesions that infiltrate the skin and cause thickening, especially on the face (known as leonine facies). Furthermore, LL is characterized by extensive bilaterally symmetric cutaneous lesions with poorly defined borders and raised indurated centers.3
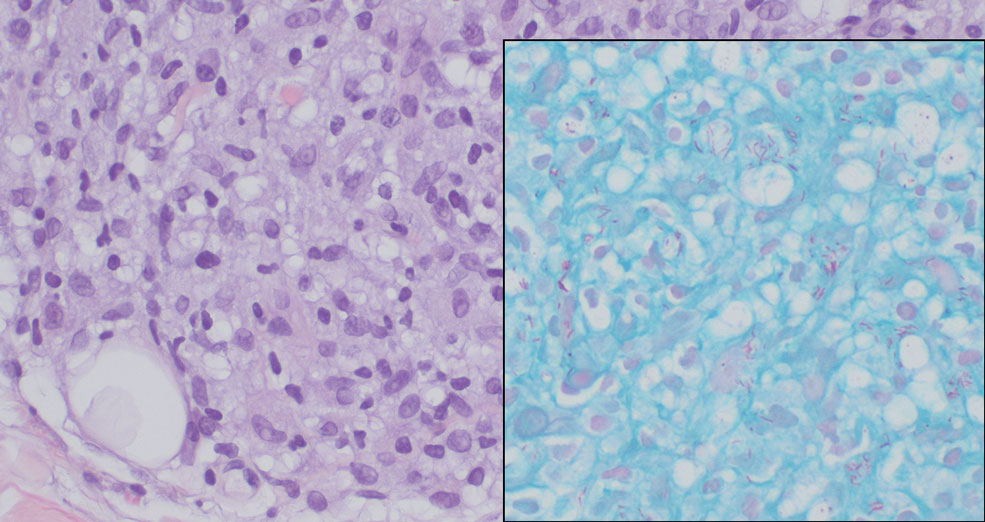
Lepromatous leprosy transmission is not fully understood but is thought to occur via airborne droplets from coughing/sneezing and nasal secretions.2 Histopathology generally shows a dense and diffuse granulomatous infiltrate that involves the dermis but is separated from the epidermis by a zone of collagen (grenz zone).3 Histology is characterized by the presence of lymphocytes and numerous foamy macrophages (lepra or Virchow cells) containing M leprae organisms. In persistent lesions, the high density of uncleared bacilli forms spherical cytoplasmic clumps known as globi within enlarged foamy histiocytes (Figure 1).4 The macrophages form granulomatous lesions in the skin and around nerve bundles, resulting in tissue damage and decreased sensation. The current standard of care for LL is a multidrug combination of dapsone, rifampin, and clofazimine. Early diagnosis and complete treatment of LL is crucial, as this approach typically leads to complete cure of the disease.
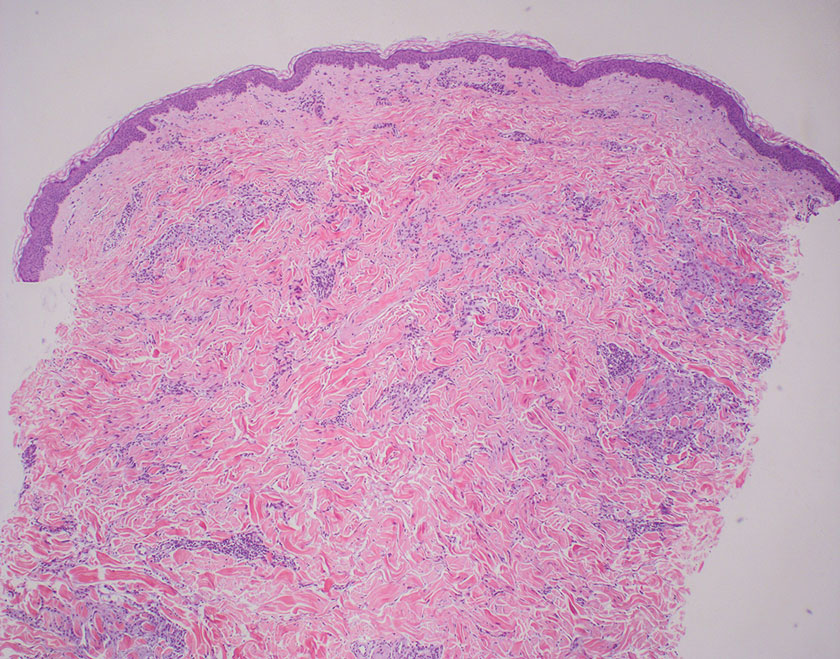
The differential diagnosis for LL includes granuloma annulare (GA), mycosis fungoides (MF), sarcoidosis, and subacute cutaneous lupus erythematosus (SCLE). Granuloma annulare is a noninfectious inflammatory granulomatous skin disease that manifests in a localized, generalized, or subcutaneous pattern. Localized GA is the most common form and manifests as self-resolving, flesh-colored or erythematous papules or plaques limited to the extremities.5,6 Generalized GA is defined by more than 10 widespread annular plaques involving the trunk and extremities and can persist for decades.6 This form can be associated with hyperlipidemia, diabetes, autoimmune disease and immunodeficiency (eg, HIV), and rarely with lymphoma or solid tumors. On histology, GA shows necrobiosis surrounded by palisading histiocytes and mucin (palisading GA) or patchy interstitial histiocytes and lymphocytes (interstitial GA)(Figure 2).6 This palisading pattern differs from the histiocytes in LL, which contain numerous acid-fast bacilli and bacterial clumps. Topical and intralesional corticosteroids are first-line therapies for GA.
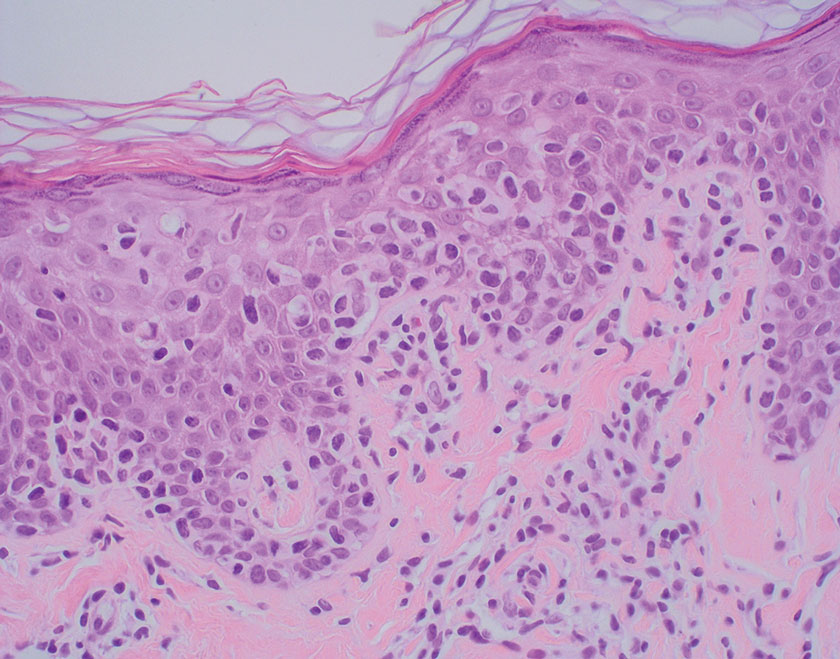
Mycosis fungoides is a cutaneous T-cell lymphoma characterized by proliferation of CD4+ T cells.7 In the early stages of MF, patients may present with multiple erythematous and scaly patches, plaques, or nodules that most commonly develop on unexposed areas of the skin, but specific variants frequently may cause lesions on the face or scalp.8 Tumors may be solitary, localized, or generalized and may be observed alongside patches and plaques or in the absence of cutaneous lesions.7 The pathologic features of MF include fibrosis of the papillary dermis, individual haloed atypical lymphocytes in the epidermis, and atypical lymphoid cells with cerebriform nuclei (Figure 3).9 Granulomatous MF is characterized by diffuse nodular and perivascular infiltrates of histiocytes with small lymphocytes without atypia, eosinophils, and plasma cells. Small lymphocytes with cerebriform nuclei and larger lymphocytes with hyperconvoluted nuclei also may be seen, in addition to multinucleated histiocytic giant cells. Although MF commonly manifests with epidermotropism, it typically is absent in granulomatous MF (GMF).10 Granulomatous MF may manifest similarly to LL. Noduloulcerative lesions and infiltration of atypical lymphocytes into the epidermis (epidermotropism) are much more common in GMF than in LL; however, although ulcerative nodules are not a common feature in patients with leprosy (except during reactional states [ie, Lucio phenomenon]) or secondary to neuropathies, they also can occur in LL.11 In GMF, the infiltrate does not follow a specific pattern, whereas LL infiltrates tend to follow a nerve distribution. Treatment for MF is determined by disease severity.12 First-line therapy includes local corticosteroids and phototherapy with UVB irradiation.
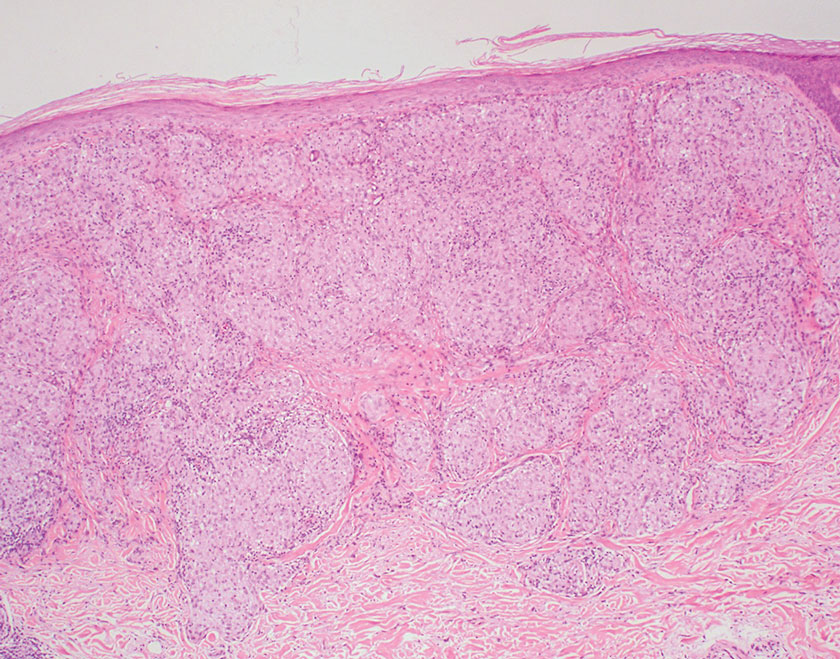
Sarcoidosis is a multisystem disease that demonstrates nonspecific clinical manifestations affecting the lungs, eyes, liver, and skin.13 Environmental exposures to silica and inorganic matter have been linked to an increased risk for sarcoidosis, with patients presenting with fatigue, fever, and arthralgia.13 Skin manifestations include subcutaneous nodules, polymorphous plaques, and erythema nodosum—nodosum—the most common cutaneous presentation of sarcoidosis. Erythema nodosum manifests as symmetrically distributed, nonulcerative, painful red nodules on the skin, especially the lower legs. The histopathology of sarcoidosis shows noncaseating granulomas with activated T-lymphocytes, epithelioid cells, and multinucleated giant cells (Figure 4). Although granulomas occur in both LL and sarcoidosis, those in sarcoidosis typically consist of epithelioid cells surrounded by a rim of lymphocytes, whereas LL granulomas contain foamy histiocytes and multinucleated giant cells. Treatment of sarcoidosis depends on disease progression and generally involves oral corticosteroids, followed by corticosteroid-sparing regimens.
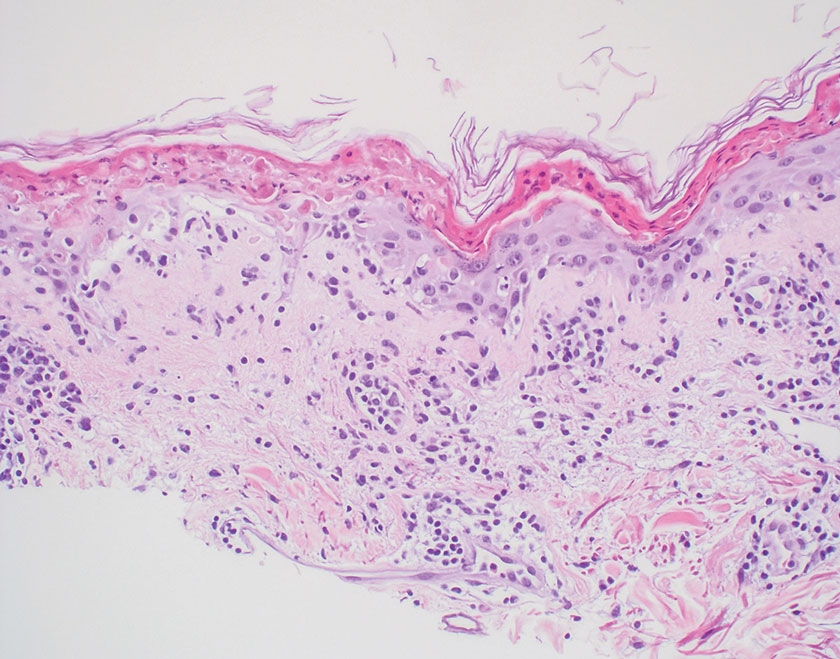
Subacute cutaneous lupus erythematosus is a chronic autoimmune disease that predominantly affects younger women. Common findings in SCLE include red scaly plaques and ring-shaped lesions on sun-exposed areas of the skin.14 Subacute cutaneous lupus erythematosus primarily is characterized by a photosensitive rash, often with arthralgia, myalgia, and/or oral ulcers; less commonly, a small percentage of patients can experience central nervous system involvement, vasculitis, or nephritis. The histologic findings of SCLE include hydropic degeneration of the basal cell layer and periadnexal infiltrates (Figure 5). The incidence of SCLE often is associated with anti-Ro (SSA) and anti-La (SSB) antibodies.15 Treatment of SCLE focuses on managing skin symptoms with corticosteroids, antimalarials, and sun protection.