Perianal Condyloma Acuminatum-like Plaque





The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
,Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
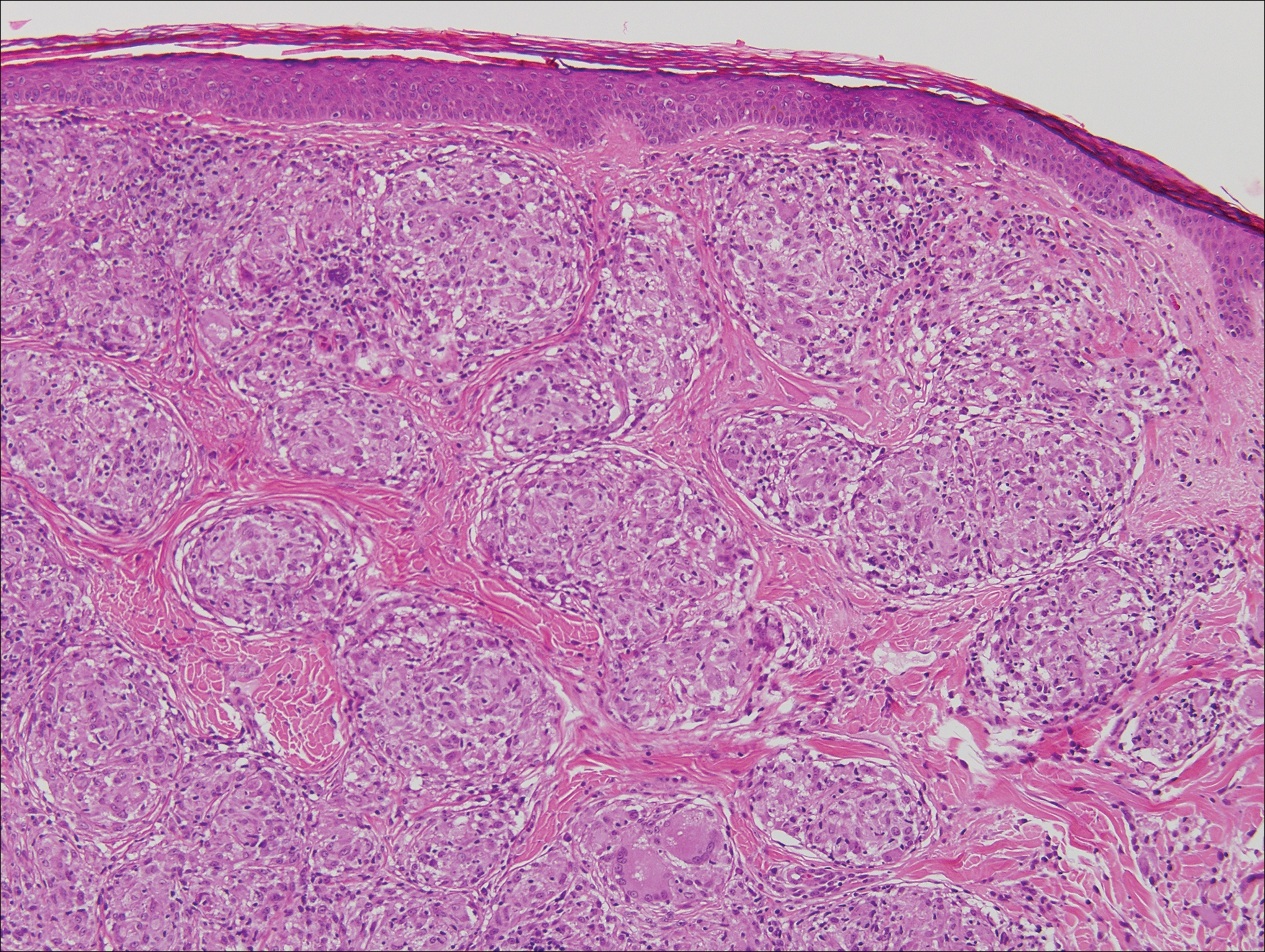
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
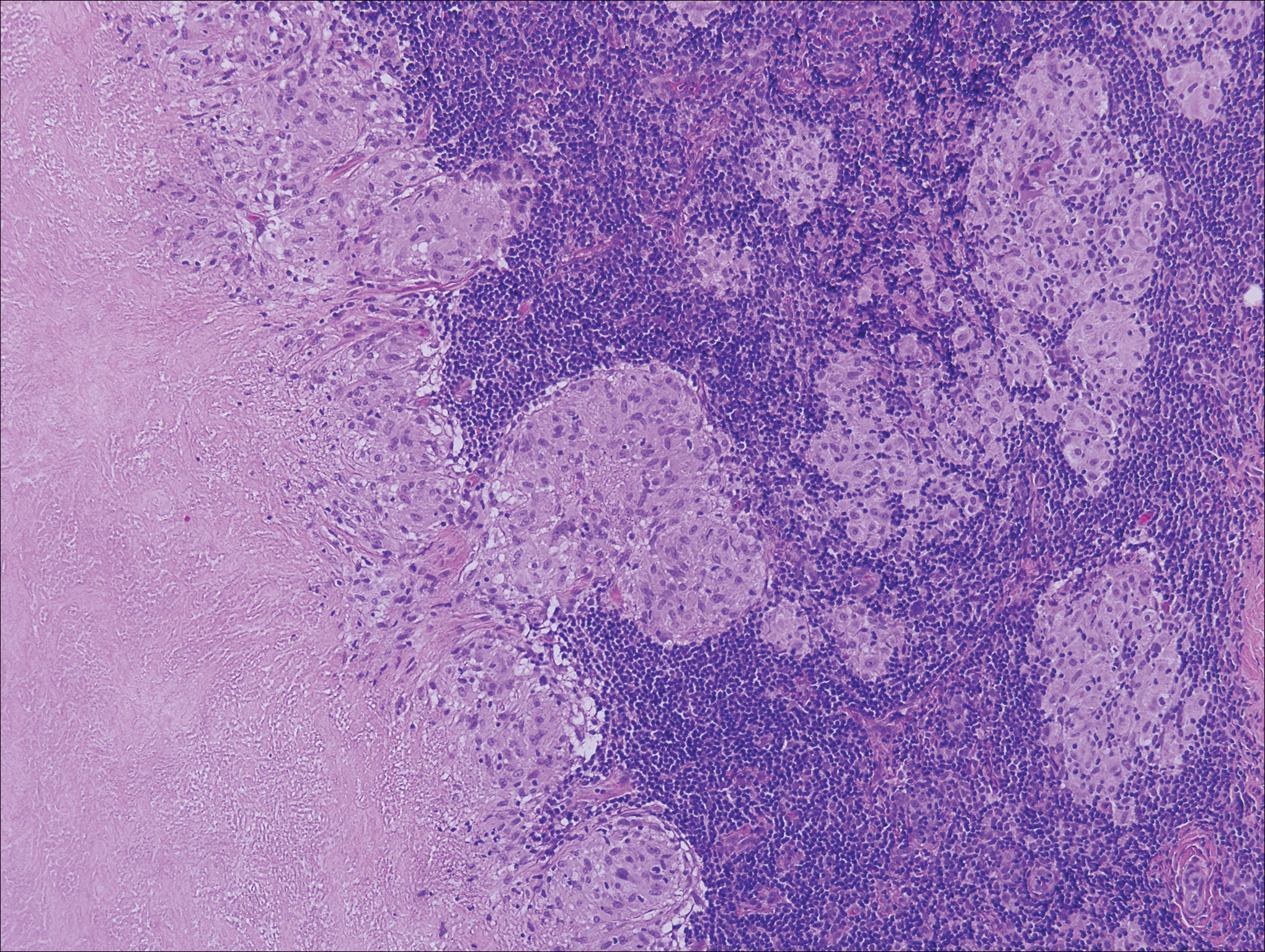
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
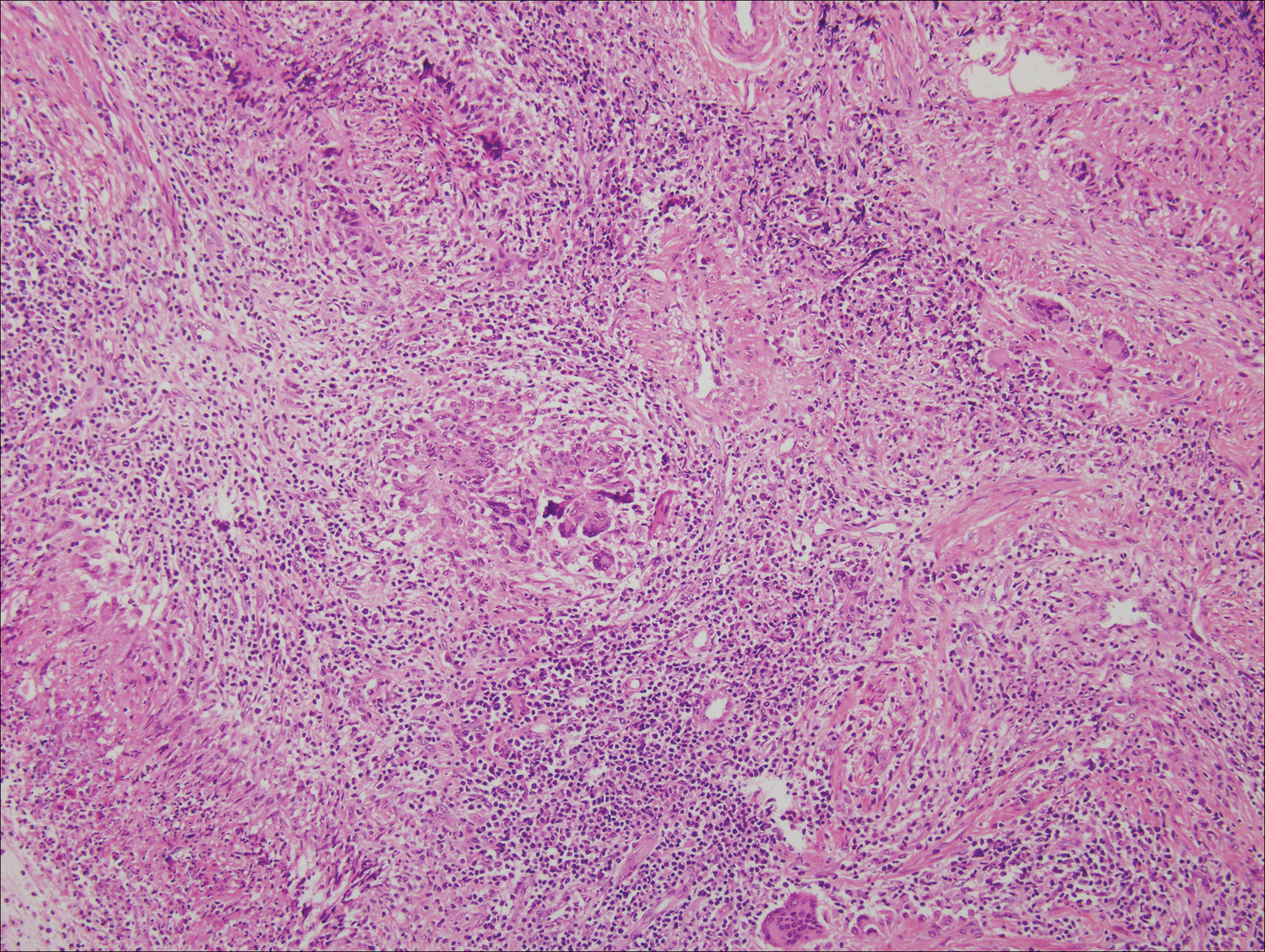
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
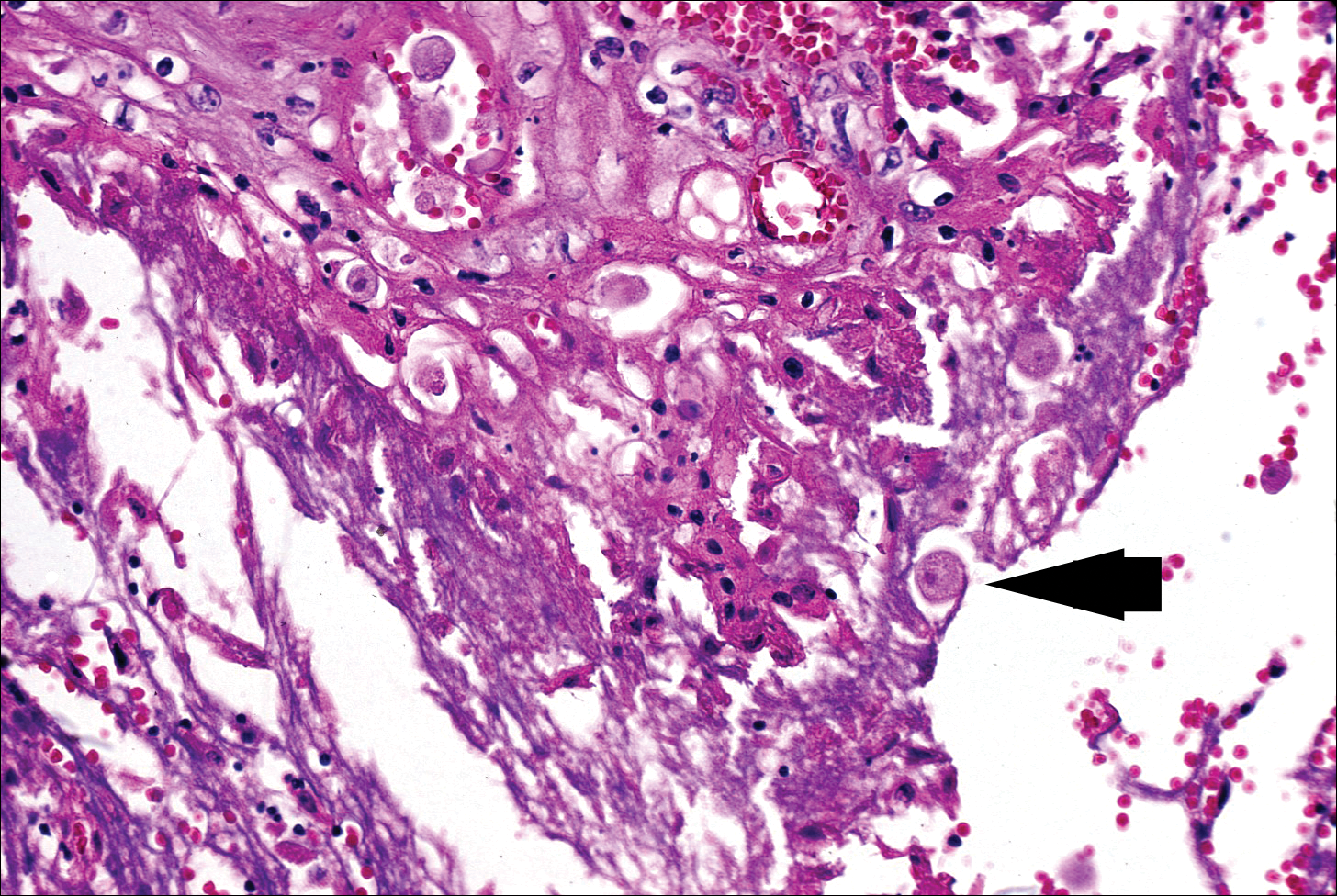
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.