Nail-Patella Syndrome: Clinical Clues for Making the Diagnosis





Nail-patella syndrome (NPS) is a rare autosomal-dominant disorder characterized by the classic triad of fingernail dysplasia, patellar absence/hypoplasia, and presence of iliac horns. We describe the various features of NPS, focusing on dermatologic and musculoskeletal findings. A 69-year-old man presented to the dermatology clinic for a routine skin cancer screening. Physical examination revealed hypoplastic fingernails with longitudinal ridging, splitting, and triangular lunulae; left patellar absence and right patellar hypoplasia; and bilateral iliac horns that had been present since birth. His medical history was remarkable for glaucoma, hypertension, osteoporosis, and chronic kidney disease. A detailed awareness of the classic findings of NPS can facilitate its early recognition and enable appropriate treatment and long-term screening.
Practice Points
- Nail-patella syndrome (NPS) is a multisystem disease.
- Nail findings (eg, triangular lunulae) may be the first clue to NPS and should prompt investigation of associated renal, ocular, neurologic, skeletal, and orthopedic abnormalities.
- Early intervention and a multidisciplinary approach to care can improve morbidity and mortality in patients with NPS.
Nail-patella syndrome (NPS), also known as hereditary osteo-onychodysplasia syndrome, is a rare autosomal-dominant disorder with an estimated incidence of 1 per 50,000 individuals in the United States. Nail-patella syndrome presents due to a heterozygous loss-of-function mutation in the LIM homeobox transcription factor 1 beta gene, LMX1B, on chromosome 9q34.1 LMX1B gene mutations are fully penetrant, but there is variable expressivity, even within families.2
Case Report
A 69-year-old man presented to the dermatology clinic for a routine skin cancer screening. The patient’s history was remarkable for dystrophic fingernails and toenails since birth. In his 20s he developed progressively worsening instability of the left knee and chronic back pain due to scoliosis, lumbar lordosis, and spinal disc herniation. Since then, he underwent knee surgery and 7 back surgeries for rheumatologic disease. His medical history also was remarkable for osteoporosis, hypertension, and glaucoma. Family history was notable for similar findings in the patient’s sister; mother; and maternal aunt, uncle, and grandmother, all with varying disease severity.
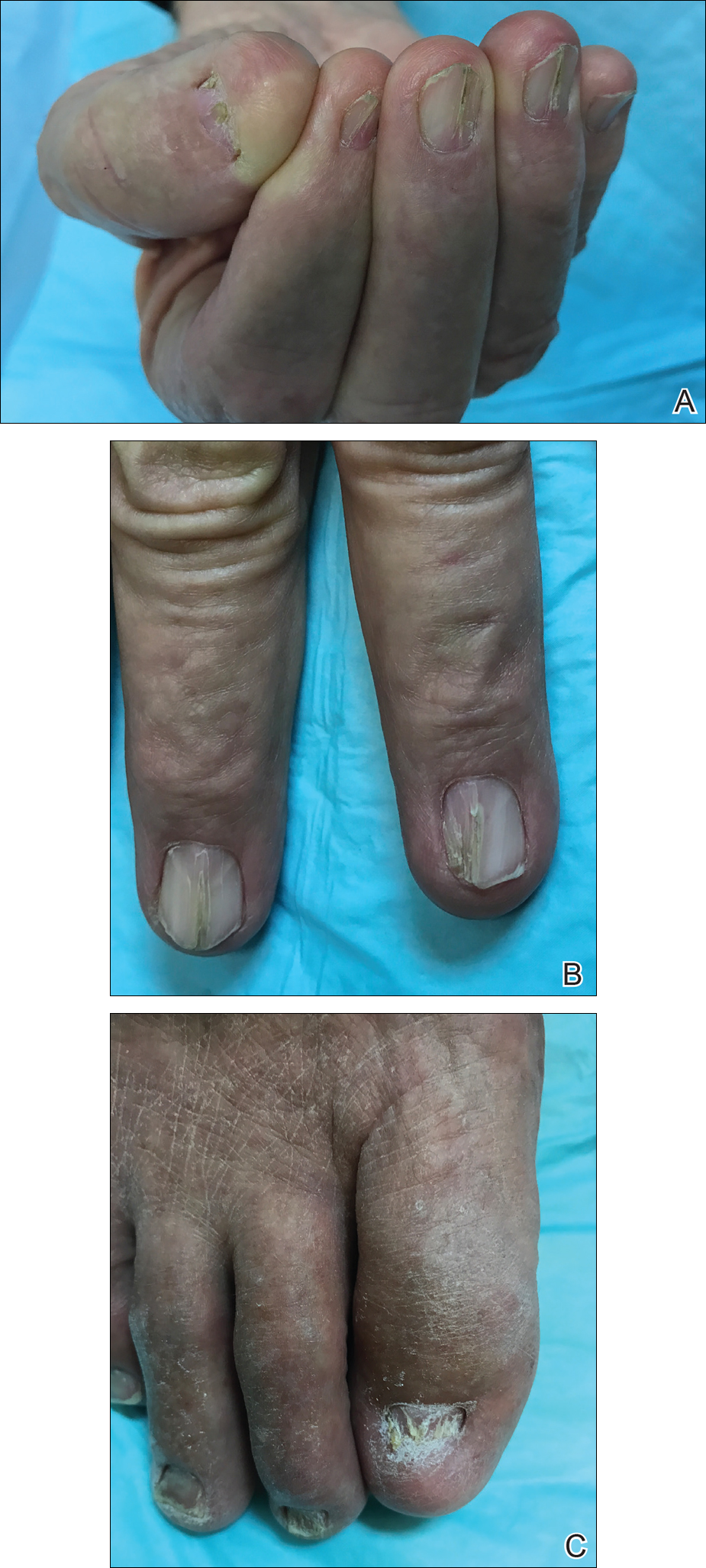
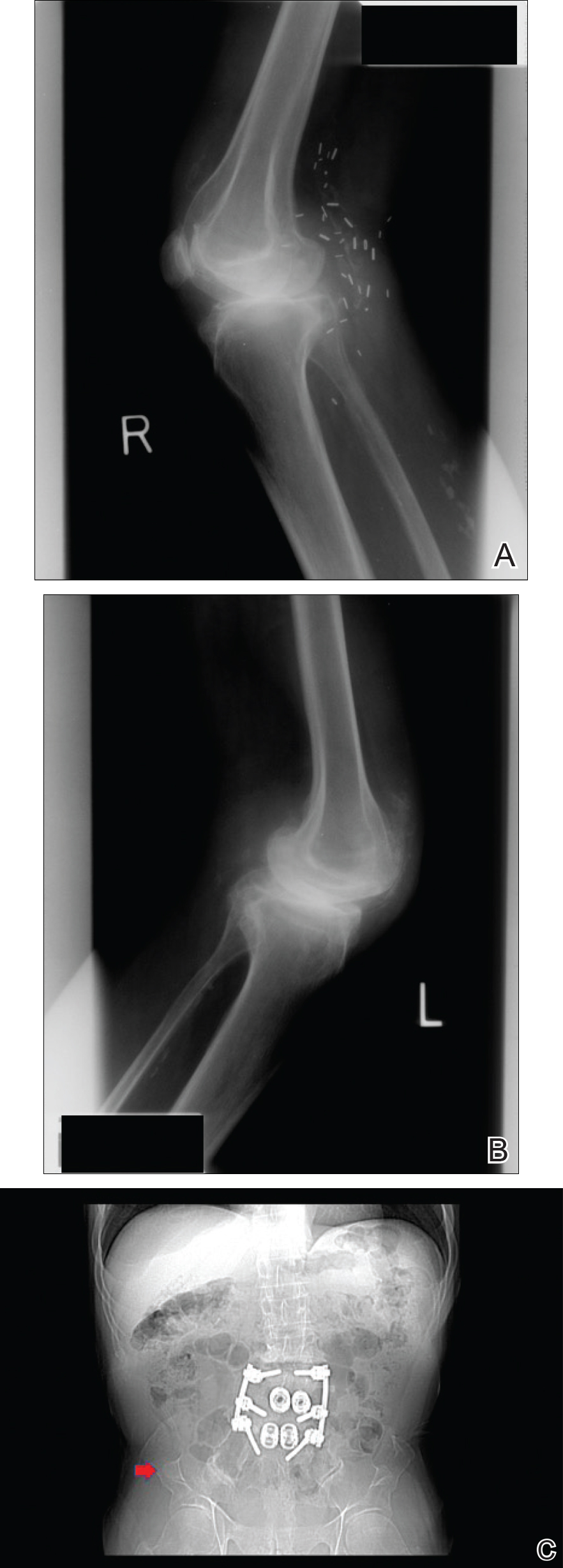
Physical examination was remarkable for bilateral fingernail hypoplasia that was most prominent on the thumb, with improvement in each nail on progression toward the fifth digit (Figure 1A). Triangular fingernail lunulae, longitudinal ridging, and nail splitting were present (Figure 1A and 1B). Hypoplastic crumbly toenails also were appreciated (Figure 1C). Skin creases over the distal interphalangeal joints of the fingers and toes were conspicuously absent. Limited range of motion was noted in multiple joints, with profound limitation of bilateral elbow extension. Review of prior imaging reports revealed bilateral iliac horns as well as left patellar absence and right patellar hypoplasia (Figure 2). Urinalysis was remarkable for proteinuria and microscopic hematuria. Given the constellation of examination findings and positive family history, a diagnosis of NPS was made.
,
Comment
Nail-patella syndrome is characterized by variable dermatologic, neurologic, nephrogenic, ophthalmologic, and orthopedic clinical manifestations.3 Almost all patients with NPS have bilateral and symmetric nail changes, including absent or hypoplastic nails with ridging, splitting, or discoloration and triangular-shaped lunulae.1,4 Nail findings are the most consistent findings of NPS, as they are present in more than 98% of patients.5 The thumb often is the most severely affected nail, with improvement appreciated on progression toward the fifth digit, as seen in our patient (Figure 1A).5 Each individual nail usually is more severely affected on its ulnar side. When toenails are involved, the abnormalities tend to be less severe, and the little toenail is most commonly affected. Distal digital changes also are observed in almost all patients. Loss of dorsal creases in the skin overlying the distal interphalangeal joints can be considered as a diagnostic clue.3,4
There are a variety of orthopedic manifestations of NPS. Hypoplastic or absent patellae leading to recurrent subluxations or dislocations is a common finding.4 Bilateral symmetric bone formations (horns) arising from the iliac crest are pathognomonic but only found on radiography 70% of the time.6 Occasionally these protuberances can be palpated on physical examination,5 though this finding was not appreciated in our patient. Dysplasia of the elbows may result in limited elbow extension and limited pronation and supination. Early degenerative arthritis, lumbar lordosis, and scoliosis also are not uncommon. In addition, skeletal integrity is compromised, leading to early osteoporosis and increased risk for fractures.5
Nephropathy develops in approximately 30% to 40% of patients and is a major determinant of mortality in these patients.2 Mutations in the LMX1B gene lead to abnormal development of podocytes and reduction in collagen in the glomerular basement membrane. The first sign of renal involvement usually is proteinuria, with or without microscopic hematuria. As in our patient, many patients develop hypertension. Patients may progress to develop nephrotic syndrome and end-stage renal failure (5%–10%).7 Death from NPS-related nephropathy has occurred, even in childhood.4,5
Primary open-angle glaucoma has been recognized as a feature of NPS.8 It is the most frequent ocular abnormality observed, followed by ocular hypertension and Lester sign of the iris.3,5 These conditions also are more common in younger patients with NPS than in the general population.5 Important neurologic findings include epilepsy, peripheral neuropathy, attention deficit disorder, major depressive disorder, and vasomotor problems.9
Our case highlights the importance of recognizing this rare condition to provide a multidisciplinary approach to care that addresses all aspects of LMX1B-associated disease in affected individuals. Nail findings may be the first clue to the need for additional screenings in these patients. Nail-patella syndrome patients should undergo thorough ophthalmologic examinations every 2 years, including measurement of intraocular pressure, examination of the optic disc, and assessment of visual fields. Given the variability in severity of joint problems and the unpredictable anatomy of the joints, magnetic resonance imaging of the joints is recommended prior to orthopedic intervention. Most importantly, physicians should recognize this genodermatosis to implement periodic screenings for renal disease, as up to 40% of NPS patients develop kidney failure. Annual blood pressure measurements, urinalysis, and measurement of the protein to creatinine ratio in the urine are recommended. For patients with end-stage renal failure, renal transplantation results in cure of nephropathy and may even result in nail regrowth.10 Further, this case is notable in that it describes a patient with NPS who is older than most other individuals presenting with the condition, thereby revealing novel information about NPS in its more advanced stages.