Detecting and managing hereditary colorectal cancer syndromes in your practice





ABSTRACTHereditary syndromes account for 5% to 10% of cases of colorectal cancer. In clinical practice, patients with these syndromes need to be identified to ensure that they and their families receive genetic counseling and testing and appropriate risk-reducing treatment. Genetic testing can offer a precise diagnosis. It allows for risk stratification and focused management and surveillance.
KEY POINTS
- Hereditary colorectal cancer syndromes carry a substantial risk of intestinal and extraintestinal tumors.
- Affected patients need increased cancer surveillance and may benefit from prophylactic surgery.
- Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history.
- Patients suspected of having hereditary colorectal cancer syndromes should be referred for genetic counseling and, if appropriate, for genetic testing.
Diagnosis of Lynch syndrome
The diagnosis of Lynch syndrome is based on molecular pathologic analysis (performed on tumor samples) and confirmed by genetic testing.
Molecular pathologic evidence of Lynch syndrome includes microsatellite instability and loss of expression of one or more of the DNA mismatch repair proteins (detected using immunohistochemistry) (more on these below). The revised Bethesda guidelines (TABLE 3) were intended to identify individuals whose tumors should be tested for one or both of these phenomena.9
In 2009, the Evaluation of Genomic Applications in Practice and Prevention working group recommended that all patients with newly diagnosed colorectal cancer undergo microsatellite instability analysis, immunohistochemistry testing, or both, regardless of whether they meet the Amsterdam II or the Bethesda guideline criteria.10
Microsatellite instability analysis. Microsatellites are short sequences of repeated DNA. The tumor cells of patients who carry defective mismatch repair genes have microsatellites that are longer or shorter than in normal cells, a condition called microsatellite instability (ie, “MSI-high”).
Microsatellite instability testing, using a standardized panel of five DNA markers, is performed on normal and tumor tissue. If more than two of the five microsatellite markers in the tumor show instability, the lesion is considered to have a high level of microsatellite instability. About 15% of colorectal cancers have this high level, although most are not associated with Lynch syndrome and lose MLH1 expression by promoter methylation.11,12
While only 2% of patients with colorectal cancer have Lynch syndrome, from 90% to 95% of colorectal cancers from patients with Lynch syndrome have high levels of microsatellite instability.10 The presence of MLH1 promoter hypermethylation, the BRAF mutation V600E, or both within the tumor suggests that the cancer is not associated with Lynch syndrome.
Some families that meet the Amsterdam I criteria have microsatellite-stable tumors: their condition has been called familial colorectal cancer type X.13 This condition is associated with a higher risk of colorectal cancer but not the other malignancies observed in Lynch syndrome.
Immunohistochemistry is performed to assess for expression of the mismatch repair proteins MSH2, MSH6, MLH1, and PMS2. Absence of expression of the specific protein within tumor cells compared with normal cells within the specimen suggests dysfunction of the specific gene and guides germline mutation testing (Figure 1). For example, a patient who lacks expression of the MSH2 protein in his or her colon cancer most likely has a mutation in the MSH2 gene. Therefore, germ-line genetic testing should initially target the MSH2 gene. Approximately 88% of Lynch syndrome-associated colorectal cancers have abnormal immunohistochemical staining.10
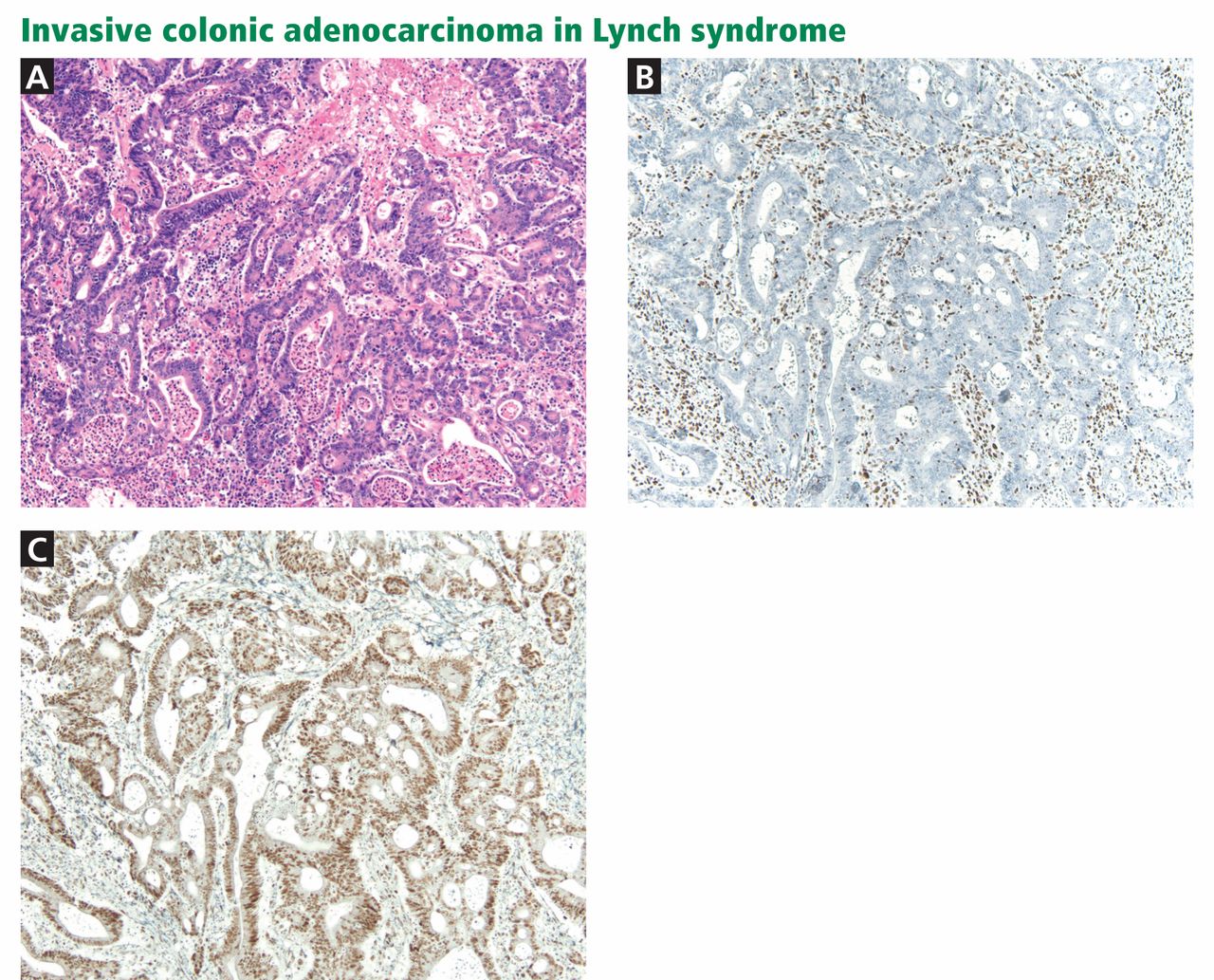
Testing for microsatellite instability and mismatch repair gene expression ideally precedes germline genetic testing and helps to guide which gene or genes should be tested.9,14
Genetic testing for Lynch syndrome is routinely performed on a blood or saliva sample, using DNA from white blood cells and sequencing the gene or genes involved to look for mutations. Positive results from a germline genetic test confirm the diagnosis of Lynch syndrome and allow for predictive testing for relatives at risk. The term Lynch syndrome is used exclusively to describe individuals with evidence of a mutation in one of the mismatch repair genes.15
If a patient’s results are positive, genetic counseling and genetic testing should be offered to at-risk relatives age 18 and over.
Management of Lynch syndrome
Aggressive cancer surveillance is essential for people with Lynch syndrome and for those who are considered at risk but have not pursued genetic testing, such as a sibling of a person with Lynch syndrome.
Colorectal cancer. Colonoscopy is recommended every 1 to 2 years beginning at the age of 20 to 25 years, or 2 to 5 years earlier than the age of the youngest relative affected with colorectal cancer if the initial diagnosis was before age 25. When patients turn 40 years old, colonoscopy is done annually.16–18 A significant reduction in cancer incidence and in the mortality rate has been shown with colonoscopic surveillance.19–21
Chemoprevention may also have a role. Patients with Lynch syndrome who took aspirin 600 mg per day for an average of 25 months had a significantly lower incidence of colorectal cancer during a 55-month follow-up period compared with patients randomized to placebo.22
For patients with Lynch syndrome who are diagnosed with colorectal cancer, the high risk of metachronous cancers after standard segmental colectomy calls for a more extended resection. Retrospective analysis of 382 Lynch syndrome patients found that none of the 50 who underwent total or subtotal colectomy were diagnosed with metachronous colorectal cancer, whereas a metachronous cancer developed in 74 (22%) of the 332 patients who had had segmented resection.2 Annual surveillance of the remaining colon, rectum, or both is indicated postoperatively.
Gynecologic cancers. Women with Lynch syndrome should also consider gynecologic surveillance and risk-reducing surgery. This includes annual gynecologic examination, transvaginal ultrasonography, and endometrial aspiration, beginning at age 30 to 35 years. Although this surveillance does detect premalignant lesions and early symptomatic cancers, its effect on the mortality rate is unknown. Hysterectomy with bilateral salpingo-oophorectomy has been shown to significantly reduce endometrial and ovarian cancers in women with Lynch syndrome.23,24
Urothelial cancers. Carriers of MSH2 mutations have a significantly higher risk of urothelial cancers.4 Therefore, MSH2 carriers should consider ultrasonography of the urinary tract, urinary cytology, and urinalysis every 1 to 2 years beginning at age 40.4
Other extracolonic cancers. Poor evidence exists for systematic screening for the other extracolonic tumors associated with Lynch syndrome. However, the National Comprehensive Cancer Network advises considering esophagogastroduodenoscopy with extended duodenoscopy as well as capsule endoscopy every 2 to 3 years beginning at age 30 to 35.14