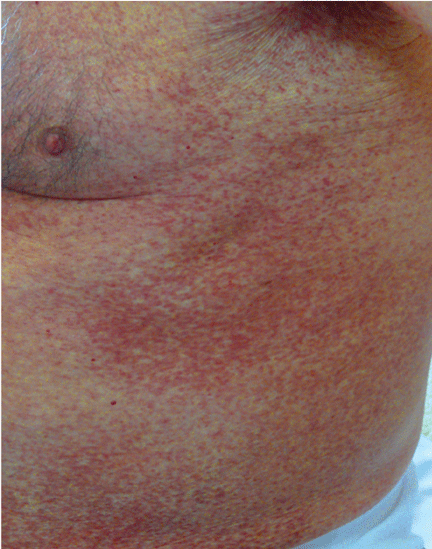
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
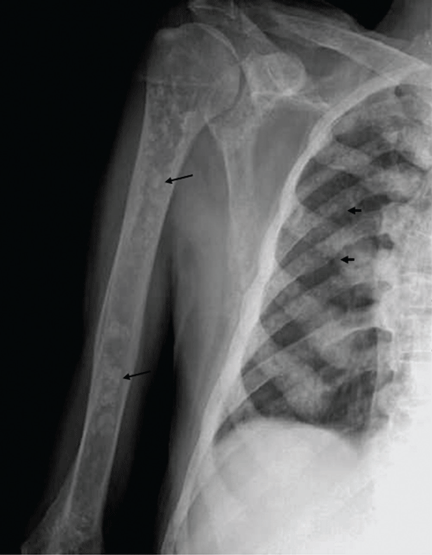
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
,
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
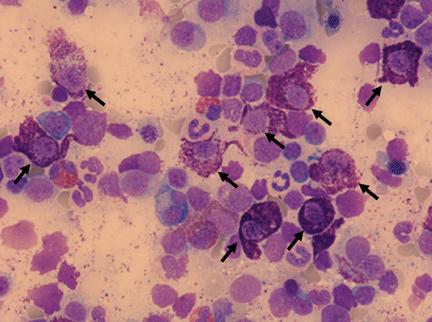
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).
Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
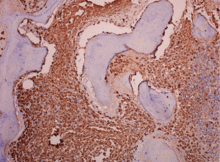
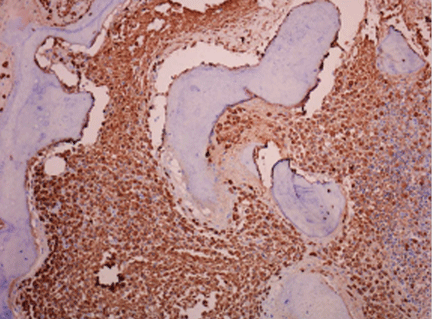
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).
One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.