A 48-year-old woman with an ecchymotic rash





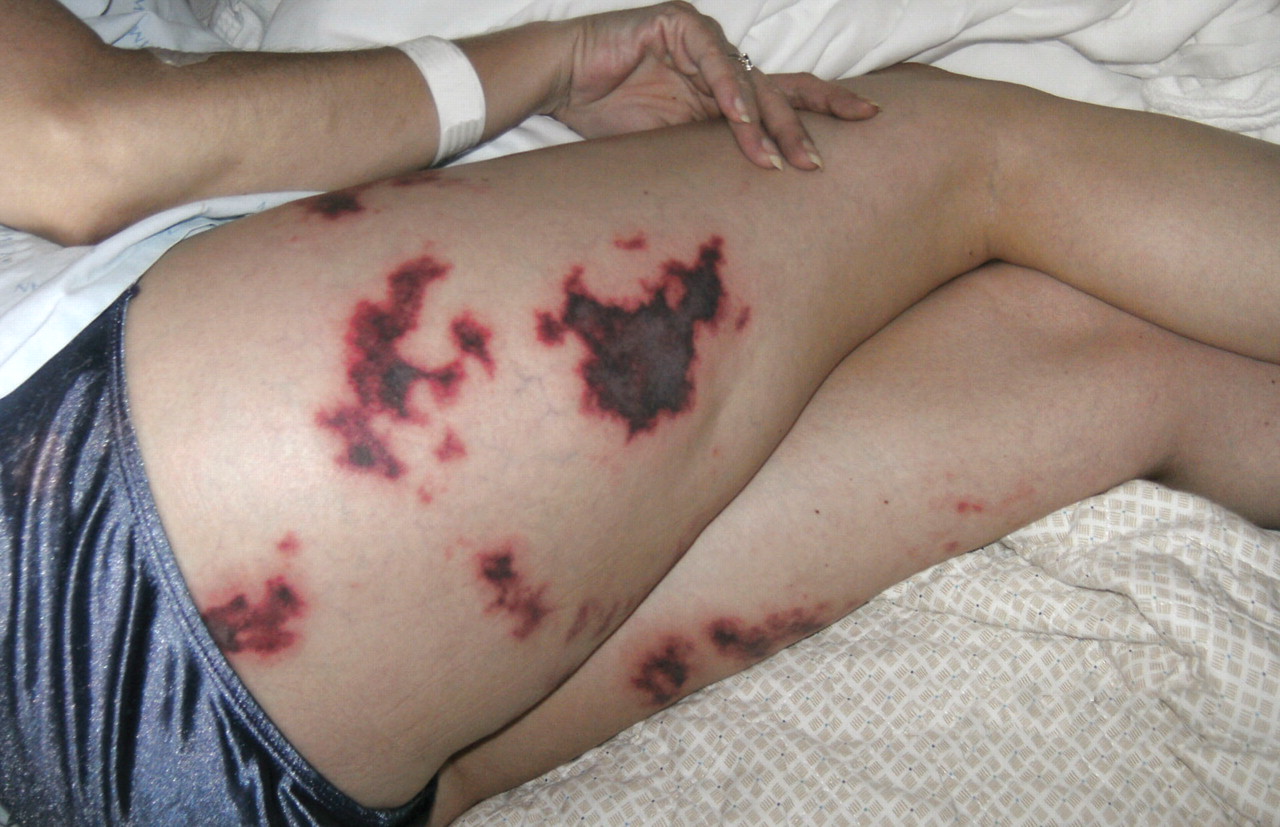
She had no constitutional symptoms and no history of venous thromboembolism, stroke, pregnancy loss, recent anticoagulation, or endovascular procedures.
Q: What is the most likely diagnosis?
,- Chronic meningococcemia
- Cholesterol embolism
- Antiphospholipid syndrome
- Cryoglobulinemic vasculitis
- Heterozygous protein C deficiency
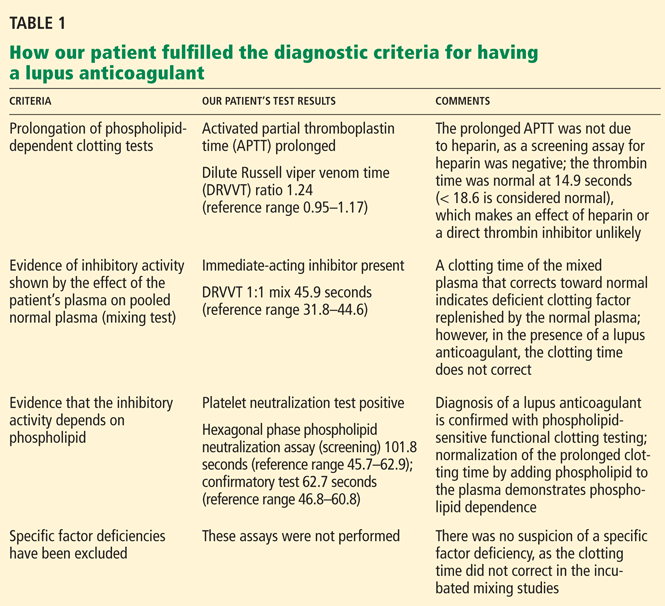
A: The most likely diagnosis is skin necrosis due to intravascular thrombosis, consistent with antiphospholipid syndrome. By clinical and laboratory criteria, the patient has systemic lupus erythematosus. Pain and swelling in multiple joints is indicative of polyarthritis associated with lupus. Retesting 12 weeks later again detected lupus anticoagulant, confirming the diagnosis of antiphospholipid syndrome.2
In the hospital, the patient was started on unfractionated heparin, later switched to warfarin. Her skin lesions gradually cleared, her pain diminished significantly, and no new lesions appeared after the start of anticoagulation therapy. For her lupus, she was started on hydroxychloroquine (Plaquenil), which has been suggested to also have an adjuvant antithrombotic role in antiphospholipid syndrome.2 On a follow-up visit 3 months later, she was doing well.
MORE ABOUT ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome is termed primary when no underlying disease is identified, and secondary when it occurs in conjunction with an autoimmune rheumatologic disease, an infection, malignancy, or certain drugs.3 It is the most common cause of acquired thrombophilia.4 Arterial or venous thromboses and recurrent miscarriages are salient clinical features.
Laboratory abnormalities include the presence of a lupus anticoagulant and anticardiolipin and beta-2-glycoprotein 1 antibodies.
Skin manifestations include livedo reticularis, purpuric macular lesions, atrophie blanche, cutaneous infarcts, ulceration, and painful nodules.5 Livedo reticularis, a violaceous, lace-like cutaneous discoloration, is the most commonly described skin lesion, present in 20% to 50% of cases.5,6 Cutaneous necrosis may involve the legs, face, and ears, or it may be generalized.6
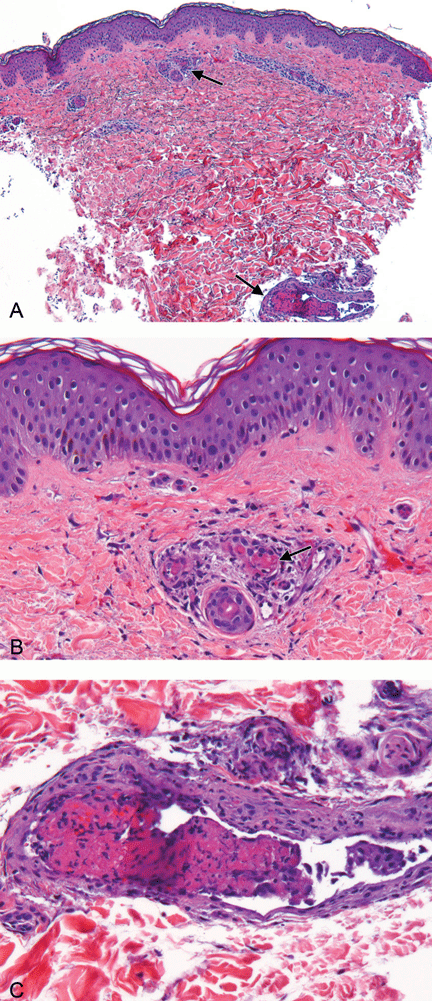
The prothrombotic state is believed to be immune-mediated, with complement activation.2 Endothelial cells and monocytes are activated by antiphospholipid antibodies with activity against beta-2-glycoprotein 1, resulting in up-regulation of tissue factor and in platelet activation.2 Histopathologic examination reveals noninflammatory vascular thromboses with endothelial damage.5
Although antiphospholipid syndrome seems to be immune-mediated, immunosuppressive therapy has not proved very effective,3 and anticoagulation is the recommended treatment.3,7
THE OTHER DIAGNOSTIC POSSIBILITIES
Chronic meningococcemia, sometimes associated with terminal complement deficiency, is associated with a petechial rash in 50% to 80% of cases. The rash can become confluent, resulting in hemorrhagic patches with central necrosis, resembling the lesions in our patient.
However, these skin lesions are due to thrombi in the dermal vessels, associated with leukocytoclastic vasculitis. These dermatopathologic changes were not seen in our patient. Moreover, meningococci were not identified in blood cultures or in the luminal thrombi and vessel walls.
Cholesterol embolism occurs when cholesterol crystals break off from severely atherosclerotic plaques, either spontaneously or after local trauma induced by angiography or aortic injury. The crystals shower downstream through the arterial system, often immediately occluding arterioles 100 to 200 μm in diameter.
Our patient had no such history, and the skin biopsy did not show the characteristic “cholesterol clefts”—biconvex, needle-shaped clefts left by the dissolved crystals of cholesterol within the occluded vessels.
Cryoglobulinemic vasculitis is an immune-complex-mediated condition involving small- to medium-size vessels, often associated with hepatitis C virus infection. Skin lesions appear in dependent areas and include erythematous macules and purpuric papules.
Cryoglobulins were not detected in our patient’s sera, nor did the skin biopsy indicate the typical leukocytoclastic vasculitis seen in this condition.
Heterozygous protein C deficiency causes venous thromboembolism and warfarin-induced skin necrosis. Spontaneous thrombosis of cutaneous arterioles (as in our patient) is not a usual manifestation. Also, our patient had normal protein C levels and no history of warfarin use before the skin lesions developed.
Acknowledgment: The authors are grateful to Dr. Judith Drazba, PhD, of Research Core Services (Imaging) at Cleveland Clinic for help in the preparation of the photomicrographs.