
Vascular signaling abnormalities in Alzheimer disease





ABSTRACT
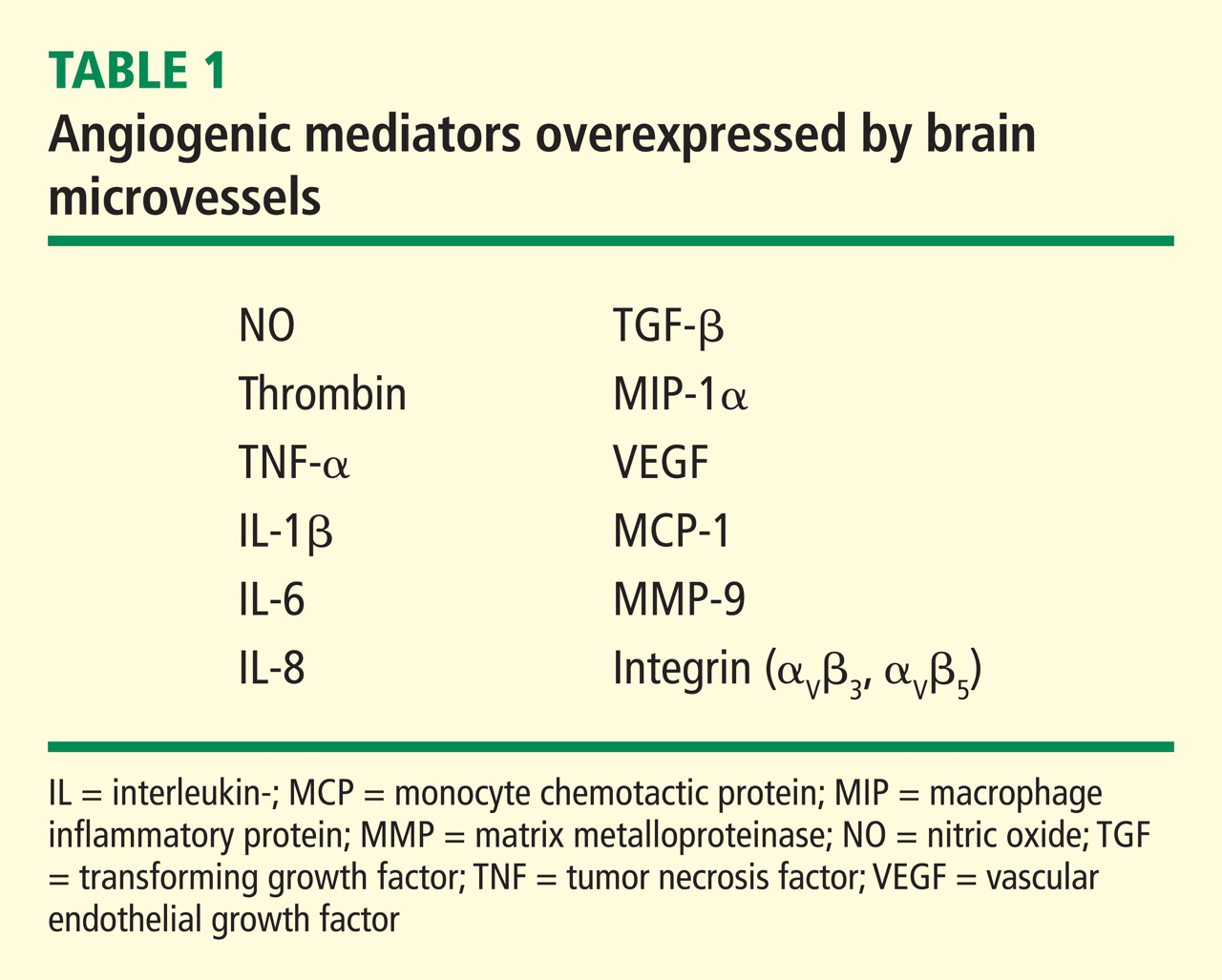
Our laboratory has documented that brain microvessels derived from patients with Alzheimer disease (AD) express or release a myriad of factors that have been implicated in vascular activation and angiogenesis. In addition, we have documented that signaling cascades associated with vascular activation and angiogenesis are upregulated in AD-derived brain microvessels. These results are consistent with emerging data suggesting that factors and processes characteristic of vascular activation and angiogenesis are found in the AD brain. Despite increases in proangiogenic factors and signals in the AD brain, however, evidence for increased vascularity in AD is lacking. Cerebral hypoperfusion/hypoxia, a potent stimulus for vascular activation and angiogenesis, triggers hypometabolic, cognitive, and degenerative changes in the brain. In our working model, hypoxia stimulates the angiogenic process; yet, there is no new vessel growth. Therefore, there are no feedback signals to shut off vascular activation, and endothelial cells become irreversibly activated. This activation results in release of a large number of proteases, inflammatory proteins, and other gene products with biologic activity that can injure or kill neurons. Pathologic activation of brain vasculature may contribute noxious mediators that lead to neuronal injury and disease processes in AD brains. This concept is supported by preliminary experiments in our laboratory, which show that pharmacologic blockade of vascular activation improves cognitive function in an animal model of AD. Thus, “vascular activation” could be a novel, unexplored therapeutic target in AD.
Alzheimer disease (AD) is a progressive, irreversible, neurodegenerative disease that affects more than 5.3 million people in the United States.1 This number is significantly higher than the previous estimate of 4.5 million and is projected to increase sharply to nearly 8 million by 2030.1 At present, the few agents that are approved by the US Food and Drug Administration for treatment of AD have demonstrated only modest effects in modifying clinical symptoms for relatively short periods; none has shown a clear effect on disease progression. New therapeutic approaches are desperately needed.
VASCULAR DISEASE AND ALZHEIMER DISEASE
Although AD is classified as a neurodegenerative dementia, there is epidemiologic and pathologic evidence of an association with vascular risk factors and vascular disease.2–6 Vascular disease appears to lower the threshold for the clinical presentation of dementia at a given level of AD-related pathology.7 The possible association of AD with vascular disease suggests that there are important pathogenic mechanisms common to both AD and vascular disease. For example, there is increasing evidence that perturbations in cerebral vascular structure and function occur in AD.8
It has been suggested that cerebral hypoperfusion/hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and contributes to the pathologic processes of AD.9 A study by Roher and colleagues reveals an association between severe circle of Willis atherosclerosis and sporadic AD.10 These observations suggest that atherosclerosis-induced brain hypoperfusion contributes to the clinical and pathologic manifestations of AD.
Hypoxia is also known to stimulate angiogenesis, especially via upregulation of hypoxia-inducible genes such as vascular endothelial growth factor (VEGF).11,12 VEGF, a critical mediator of angiogenesis, is present in the AD brain in the walls of intra-parenchymal vessels, in diffuse perivascular deposits, and in clusters of reactive astrocytes.13 In addition, intrathecal levels of VEGF in AD are related to clinical severity and intrathecal levels of amyloid-beta (Aβ).14 Emerging data support the idea that factors and processes characteristic of angiogenesis are found in the AD brain.15,16
ENDOTHELIAL ACTIVATION AND ANGIOGENESIS
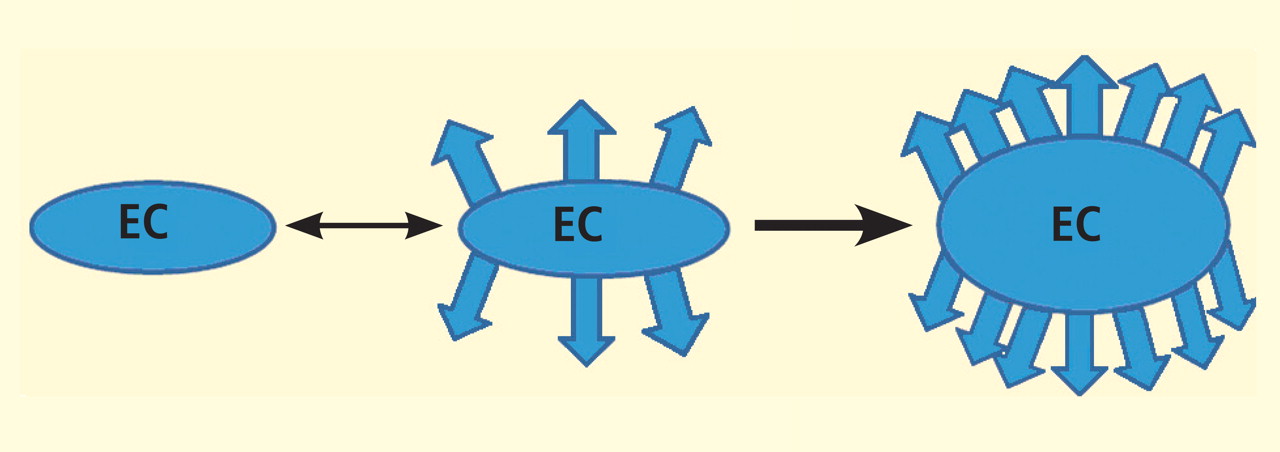
The angiogenic process is complex and involves several discrete steps, such as endothelial activation, extracellular matrix degradation, proliferation and migration of endothelial cells, and morphologic differentiation of endothelial cells to form tubes. Stimuli known to initiate angiogenesis, including hypoxia, inflammation, and mechanical factors such as shear stress and stretch,23 either directly or indirectly activate endothelial cells. Activated endothelial cells elaborate adhesion molecules, cytokines and chemokines, growth factors, vasoactive molecules, major histocompatibility complex molecules, procoagulant and anticoagulant moieties, and a variety of other gene products with biologic activity.24 The activated endothelium exerts direct local effects by producing at least 20 paracrine factors that act on adjacent cells.25
ANGIOGENIC SIGNALING MECHANISMS IN BRAIN MICROVESSELS
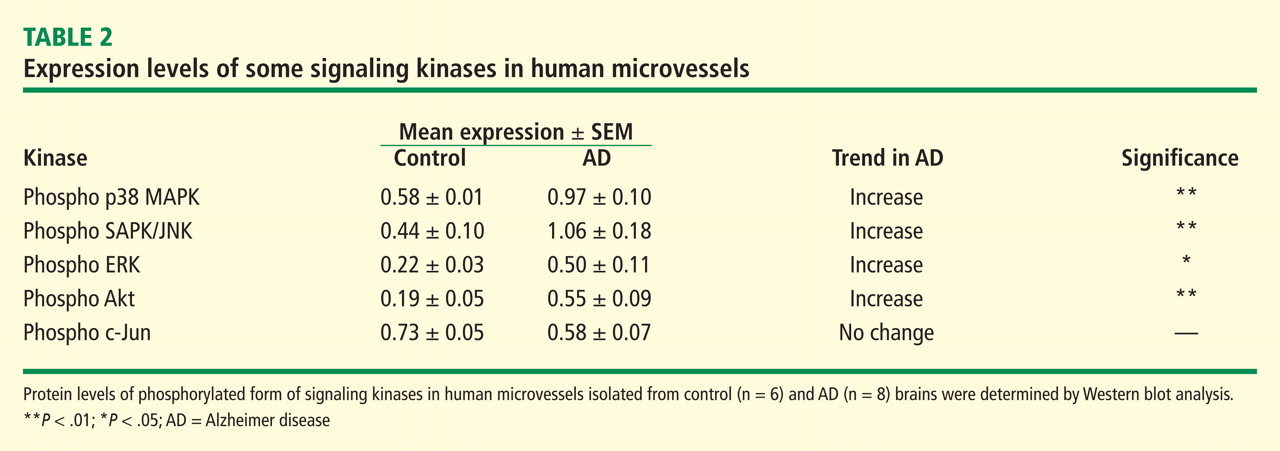
Signaling mechanisms that have been identified as important to endothelial cell viability and angiogenesis include PI3K/Akt, p38 kinase, ERK, and JNK. In this regard, intracellular Aβ accumulation is toxic to endothelial cells and decreases PI3K/Akt.26 Extracellular Aβ peptides decrease phosphorylation and thus activation of ERK and p38 kinase.26 VEGF promotes endothelial survival, proliferation, and migration through numerous pathways, including activation of ERK, p38 kinase, JNK, and Rho GTPase family members.23
VASCULAR ACTIVATION IN ALZHEIMER DISEASE
Despite increases in several proangiogenic factors in the AD brain, evidence for increased vascularity in AD is lacking. On the contrary, it has been suggested that the angiogenic process is delayed or impaired in aged tissues, with several studies showing decreased microvascular density in the AD brain.30–33 Paris et al showed that wild-type Aβ peptides have antiangiogenic effects in vitro and in vivo.34
How can the data showing antiangiogenic effects of Aβ be reconciled with the presence or expression of a large number of proangiogenic proteins by brain microvessels in AD? These conflicting observations suggest an imbalance between proangiogenic and antiangiogeneic processes in the AD brain.
Preliminary experiments in our laboratory show that pharmacologic blockade of vascular activation improves cognitive function in an animal model of AD. Thus, “vascular activation” could be a novel, unexplored therapeutic target in AD.
Acknowledgment
The authors gratefully acknowledge the secretarial assistance of Terri Stahl.
