The eyes: A window into the past





A 34-year-old woman living in southern California presents for a routine physical examination. During the eye examination, the physician notices “spots” on the retina and refers the patient to a retinal specialist.
The patient has no complaints about her vision. She has myopia (−4.5 diopters), corrected with glasses. She has no family history of ocular disease. Her medical history is unremarkable, and she is taking no medications.
THE MOST LIKELY DIAGNOSIS
,The lesions raise the suspicion of histoplasmosis, but since the patient has no other evidence of histoplasmosis, the likely diagnosis is presumed ocular histoplasmosis syndrome (POHS).
Further questioning reveals that the woman grew up on a farm in the Ohio River valley, one of two areas in the United States where Histoplasma capsulatum is highly endemic.1 (The other area is the Mississippi River valley.)
As this case shows, POHS is important to consider, especially in areas where H capsulatum is not endemic, to avert a lengthy workup for other causes of retinal lesions. It also shows the importance of a thorough history, including previous residences and travel.2
Pathogenesis is uncertain
In histoplasmosis, the infection is acquired by inhalation of microconidia of H capsulatum, usually via disruption of the soil (as in farming) and especially in areas where there are bird roosts. Infection is often asymptomatic, and fewer than 1% of people exposed develop a clinical illness 7 to 21 days after exposure.3
In disseminated histoplasmosis, eye involvement manifests as panophthalmitis or uveitis, caused by yeast implantation. The finding of eye lesions typical of histoplasmosis but in the absence of signs of disseminated histoplasmosis— as in POHS—is much more common, seen in perhaps 1% to 5% of residents in highly endemic areas.4
The pathogenesis of POHS and its association with histoplasmosis are still unclear.4–7 Some have proposed a cellular immune response to deposited fungal antigens. Others contend that patients with specific human leukocyte antigen types (eg, B7, DR2) may be susceptible.4H capsulatum DNA has been isolated in one case of POHS,6 and the classic ocular lesions are prevalent in people who live in the Ohio River valley.7 Therefore, even though a definitive causative relationship between H capsulatum exposure and POHS has not been proven, the ocular lesions are presumed to be the result of previous exposure to H capsulatum, as in this case.
Establishing the diagnosis of POHS
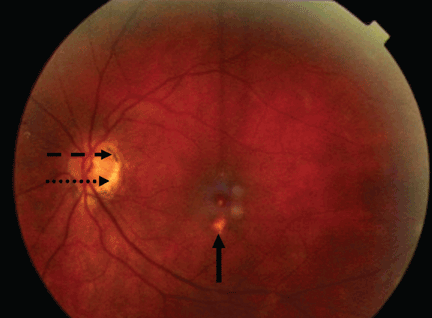
Most cases of POHS are detected on routine eye examination. The diagnosis is confirmed by a dilated eye examination showing peripheral, punched-out, atrophic scars (“histospots”), which represent focal defects in the Bruch membrane, along with a history of living in an area endemic for H capsulatum. Histo-spots range from 0.2 to 0.7 disk diameters and can occur as single or multiple lesions.8 Most often, both eyes are involved, albeit asymmetrically.9 Some areas may contain pigment deposits, as in this case.8 Peripapillary atrophy, a thinning of the retina immediately surrounding the head of the optic nerve, is also characteristic of POHS. Active inflammation in the anterior chamber and vitreous are absent.
Although most patients with POHS do not have a documented diagnosis of previous clinical Histoplasma infection, they may have a positive histoplasmin skin test, as well as lung, liver, or spleen calcifications. However, skin testing is not recommended as it may exacerbate POHS,9 and serologic testing is usually negative.10
POHS has most commonly been diagnosed in whites ages 20 to 50 (mean age 35). Men and women appear to be equally affected.
The potential for vision loss
Very few patients with ophthalmoscopic evidence of POHS develop visual symptoms.7 Still, there is a risk of choroidal neovascularization at the site of the choroidal scars. These new vessels can hemorrhage, causing impaired central vision (distorted vision, blind spots).
The trigger for choroidal neovascularization is unknown; exposure to fungal antigens and eye surgery such as LASIK have been proposed. 11 Choroidal neovascularization usually occurs 10 to 20 years after scar formation and occurs in fewer than 5% of POHS patients.10,12
HOW THE PATIENT WAS MANAGED
Given that the patient had POHS with no evidence of neovascularization, she was followed with serial visual assessments using an Amsler grid.11 For POHS with choroidal neovascularization, treatment focuses on reducing the risk of vascular complications and includes oral corticosteroids, intravitreal corticosteroid injections, laser photocoagulation, and photodynamic therapy with verteporfin (Visudyne). 4,10,13–15 Antifungal treatment is not useful, as the lesions are not proven to be caused by active infection.10
Future treatments may include antiangiogenic drugs and gene therapy.9
Since her diagnosis, the patient’s visual tests have been stable, with no neovascularization.