Lesions on the hands, high aminotransferase levels





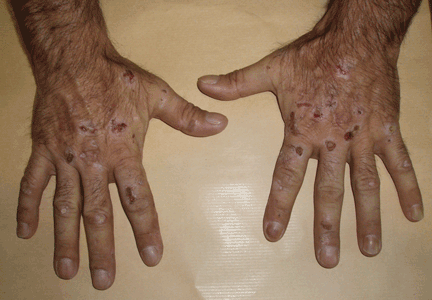
Q: Which is the most likely diagnosis?
- Addison disease
- Lupus erythematosus
- Polymorphous light eruption
- Porphyria cutanea tarda
- Bullous pemphigoid
A: Urine testing, including examination under ultraviolet light with a Wood lamp, indicates porphyria cutanea tarda. This is the most common porphyria, occurring mainly in men. Its true prevalence is not known but is estimated to be from 1:5,000 to 1:25,000.1
There are three types of porphyria cutanea tarda. About 80% of cases are type I, also referred to as “sporadic.” In type I, levels of uroporphyrinogen decarboxylase (UROD) in red blood cells are normal, but are low in the liver during episodes of the disease. In type II, UROD levels are about 50% below normal in all tissues. Type III is similar to type I, except that it occurs in more than one family member.
,The genetic mutation that produces a deficiency of UROD leads to an excess of uroporphyrins and porphyrins that are partially decarboxylated and that irreversibly oxidize. When they are deposited in the skin and the skin is exposed to the sun, they cause the classic cutaneous manifestations.1
Risk factors2 for porphyria cutanea tarda can be extrinsic (eg, high iron blood levels,2,3 excessive ethanol intake, hepatitis C,2,4 human immunodeficiency virus, estrogen use,5 dialysis for end-stage renal disease) or intrinsic (altered iron metabolism or cytochrome P450 function2).
CLINICAL PRESENTATION
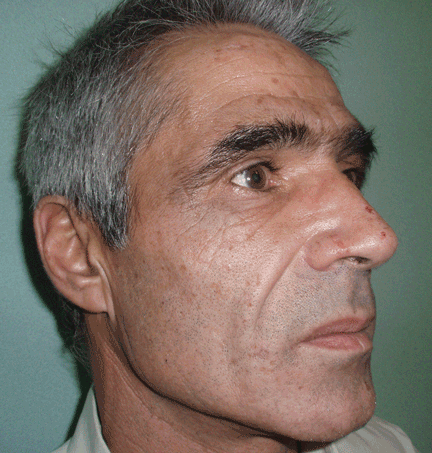
The cardinal symptom of porphyria cutanea tarda is photosensitivity, with the development of chronic blistering lesions on sun-exposed areas such as the hands, face, and forearms. Fluid-filled vesicles develop and rupture easily, and the denuded areas become crusted and heal slowly.5 Secondary infections can occur. Previous areas of blisters may appear atrophic, brownish, or violaceous. Small white plaques (milia) are also common and may precede or follow vesicle formation. These cutaneous lesions, however, are not specific to porphyria cutanea tarda and can appear in variegated porphyria and coproporphyria. Hypertrichosis5 and hyperpigmentation are usually present, mainly over the cheekbones and around the eyes. Patches of alopecia and hypopigmented sclerodermiform lesions may also be observed.
Porphyria cutanea tarda is usually accompanied by alterations in liver metabolism, affecting mainly aminotransferases and gammaglutamyltransferase. The absence of hepatitis C infection does not rule out porphyria cutanea tarda. About 50% of patients have pathologic structural changes in the liver such as lobular necrosis or fibrotic tracts, and 15% of patients have cirrhosis at presentation.6 The risk of hepatocellular carcinoma is clearly increased.6 Hepatitis C virus infection, iron overload, and excessive ethanol intake lead to a more severe liver disease.
DIAGNOSIS
The diagnosis of porphyria cutanea tarda is strongly suggested by the characteristic skin lesions in sun-exposed areas, but confirmation requires demonstration of high levels of uroporphyrins or coproporphyrins, or both.
Porphyrins accumulate in the liver, plasma, urine, and feces. Plasma porphyrin levels in porphyria cutanea tarda are usually above 10 μg/dL (normal < 1.4 μg/dL), and plasma fluorescence scanning usually shows a maximum fluorescence emission at an excitation wavelength of 619 nm. In this patient, however, the definitive diagnosis was made by chromatographic separation and the quantification of porphyrins in the urine and feces, which showed a predominance of uroporphyrins and heptacarboxyporphyrins in the urine and an excess of isocoproporphyrins in the feces.1
Analysis of UROD activity in erythrocytes can help determine the type of porphyria cutanea tarda. Type I and type III and porphyria cutanea tarda secondary to hepatotoxin exposure have normal levels, whereas type II and the hepatoerythropoietic form have abnormally low levels. Examination of the urine with a Wood lamp reveals coral pink fluorescence due to elimination of porphyrins, and this is another diagnostic clue.
Conditions that need to be ruled out include viral infection with hepatitis B or C or human immunodeficiency virus, iron overload, and hereditary hemochromatosis. Serum alpha fetoprotein level assessment, liver ultrasonography, or even biopsy may be indicated to exclude hepatocellular carcinoma.
TREATMENT
Once secondary causes of porphyria are excluded or treated (eg, advising the patient to avoid alcohol, discontinuing estrogens or iron intake), the next step in management is to reduce the patient’s porphyrin and iron loads. Phlebotomy is the standard way to reduce stores of iron throughout the body and particularly in the liver. It works by interrupting iron-mediated oxidative inhibition of hepatic UROD and the oxidation of hepatic porphyrinogens to porphyrinogens.
This adjustment must be gradual, with about 450 mL of blood removed at intervals of 1 to 2 weeks.7 This improves the cutaneous symptoms progressively, with resolution of vesicles in 2 to 3 months, improvement of skin fragility in 6 to 9 months, and normalization of porphyrin levels in 13 months. The scleroderma, atrophy, hyperpigmentation, and hypertrichosis respond more slowly and may take years to resolve.
Porphyria cutanea tarda can recur, usually with new exposure to risk factors. Treatment by phlebotomy may be stopped when the serum ferritin level has reached low-normal levels; the porphyrin levels may not yet be normal at that point but may continue to decline without additional phlebotomy sessions.
If phlebotomy is contraindicated, alternatives include iron chelation with deferoxamine (Desferal),7 or a low dose of chloroquine (Aralen) (125–250 mg orally twice a week) or hydroxychloroquine (Plaquenil) (100 mg orally twice a week) to avoid acute hepatic damage that may be caused by the release of large amounts of porphyrins that accompany standard dosing levels.