Multiple linear subcutaneous nodules





A 34-year-old woman sought consultation at our clinic for an asymptomatic swelling on her right foot that had been growing very slowly over the last 15 years. She said she had presented to other healthcare facilities, but no diagnosis had been made and no treatment had been offered.
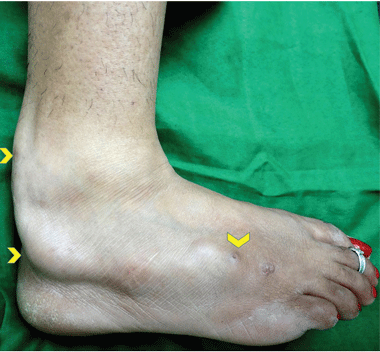
Examination revealed a linear swelling extending from the lower third to the mid-dorsal surface of the right foot (Figure 1). Palpation revealed multiple, closely set nodules arranged in a linear fashion. This finding along with the history raised the suspicion of neurofibroma and other conditions in the differential diagnosis, eg, pure neuritic Hansen disease, phaeohyphomycosis, and palisaded neutrophilic granulomatous dermatitis. The rest of the mucocutaneous examination results were normal. No café-au-lait spots, axillary freckling, or other swelling suggestive of neurofibroma was seen. She had no family history of mucocutaneous disease or other systemic disorder.
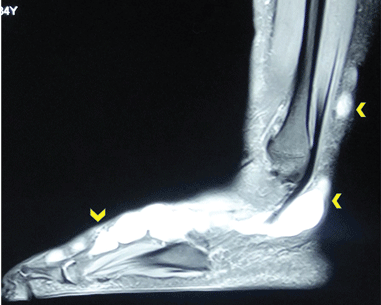
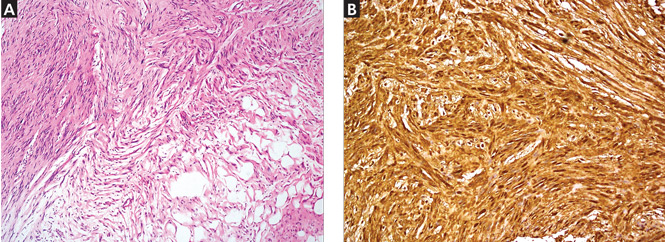
Because of the suspicion of neurofibromatosis, slit-lamp examination of the eyes was done to rule out Lisch nodules, a common feature of neurofibromatosis; the results were normal. Plain radiography of the right foot showed only soft-tissue swelling. Magnetic resonance imaging with contrast, done to determine the extent of the lesions, revealed multiple dumbbell-shaped lesions with homogeneous enhancement (Figure 2). Histopathologic study of a biopsy specimen of the lesions showed tumor cells in the dermis. The cells were long, with elongated nuclei with pointed ends, arranged in long and short fascicles—an appearance characteristic of neurofibroma. Areas of hypocellularity and hypercellularity were seen, and on S100 protein immunostaining, the tumor cells showed strong nuclear and cytoplasmic positivity (Figure 3).
,
The histologic evaluation confirmed neurofibroma. The specific diagnosis of sporadic solitary neurofibroma was made based on the onset of the lesions, the number of lesions (one in this patient), and the absence of features suggestive of neurofibromatosis.
SPORADIC SOLITARY NEUROFIBROMA
Neurofibroma is a common tumor of the peripheral nerve sheath and, when present with features such as café-au-lait spots, axillary freckling, and characteristic bone changes, it is pathognomic of neurofibromatosis type 1.1 But solitary neurofibromas can occur sporadically in the absence of other features of neurofibromatosis.
Sporadic solitary neurofibroma arises from small nerves, is benign in nature, and carries a lower rate of malignant transformation than its counterpart that occurs in the setting of neurofibromatosis.2 Though sporadic solitary neurofibroma can occur in any part of the body, it is commonly seen on the head and neck, and occasionally on the presacral and parasacral space, thigh, intrascrotal area,3 the ankle and foot,4,5 and the subungual region.6 A series of 397 peripheral neural sheath tumors examined over 30 years showed 55 sporadic solitary neurofibromas occurring in the brachial plexus region, 45 in the upper extremities, 10 in the pelvic plexus, and 31 in the lower extremities.7
Management of sporadic solitary neurofibroma depends on the patient’s discomfort. For asymptomatic lesions, serial observation is all that is required. Complete surgical excision including the parent nerve is the treatment for large lesions. More research is needed to define the potential role of drugs such as pirfenidone and tipifarnib.
THE DIFFERENTIAL DIAGNOSIS
Sporadic solitary neurofibroma can masquerade as pure neuritic Hansen disease (leprosy), phaeohyphomycosis, and palisaded neutrophilic granulomatous dermatitis. The absence of neural symptoms and no evidence of trophic changes exclude pure neuritic Hansen disease. Phaeohyphomycosis clinically presents as a single cyst that may evolve into pigmented plaques,8 and the diagnosis relies on the presence of fungus in tissue. The absence of cystic changes clinically and fungi histopathologically in this patient did not favor phaeohyphomycosis. Palisaded neutrophilic granulomatous dermatitis is characterized clinically by cordlike skin lesions (the “rope sign”) and is accompanied by extracutaneous, mostly articular features. Histopathologically, it shows intense neutrophilic infiltrate and interstitial histiocytic infiltrate along with collagen degeneration. The absence of extracutaneous and classical histologic features negated this possibility in this patient.
Though sporotrichosis and cutaneous atypical mycobacterial infections may present in linear fashion following the course of the lymphatic vessels, the absence of epidermal changes after a disease course of 15 years and the absence of granulomatous infiltrate in histopathology excluded these possibilities in this patient.
The patient was referred to a plastic surgeon, and the lesions were successfully resected. She did not return for additional review after that.