
Colorectal Carcinoma and Emerging Targeted Therapies





Colorectal cancer (CRC) is the third most common cancer and the third leading cause of cancer death in the U.S.1-3 Only 40% of cases are diagnosed in a localized stage with an estimated 5-year survival of 90%, whereas 20% of cases present with metastatic disease with a 5-year survival of about 12.5%.1 With recent advances in cancer genetics and immunology as well as approval of targeted agents by the FDA, different treatment options are now available, even for progressive disease.
This article presents a brief review of CRC with a special focus on targeted therapies in metastatic CRC (mCRC). Colorectal cancers exhibit certain mutations, which affect the tumor responsiveness to various treatment options. This article describes the role of targeted therapies in various well-established mutations.
Epidemiology
The most common tumor location is the proximal colon (42%), followed by the rectum (28%). More than 90% of patients with CRCs are aged > 50 years at diagnosis.
Women have a higher percentage of proximal tumors compared with men (46% vs 38%) and a lower percentage of rectal tumors (24% vs 31%).Among both sexes, incidence and mortality are highest in African Americans and lowest in Asian/Pacific Islanders. The estimated 5-year survival is slightly higher for rectal cancer (66.5%) than for colon cancer (64.2%), although the stage-specific survival is similar. The difference in 5-year overall survival (OS) is attributed to the higher percentage of rectal tumors diagnosed at a localized stage (44% vs 38%). Patients aged < 65 years have higher 5-year survival rates than do those aged 65 years (68.9% vs 62.0%).1
Risk Factors
Like most human cancers, multiple genetic and environmental factors are believed to play a role in the development of colorectal carcinomas, with environmental factors playing the dominant role.4
Environmental risk factors include a low-fiber diet,5 red and processed meat intake,6 a high-fat diet,7 smoking,8 heavy alcohol consumption,9 obesity,10 physical inactivity,11 alteration in intestinal flora,12 and chronic inflammation.13 Aspirin (at doses > 300 mg/d), nonsteroidal anti-inflammatory drugs, and folic acid are believed to protect against colon cancer.14
Some of the genetic factors involved in colorectal cancers include (1) the loss of tumor suppressor genes, such as APC (most common tumor suppressor mutation), p53, SMAD4 pathways, or TGF- ß pathways; (2) DNA mismatch repair defects: mutations in MLH1, MSH2 in hereditary nonpolyposis colon cancer or methylation of MLH1 in sporadic cases; and (3) CpG island methylation (CIMP pathway), methylation of MLH1, MINT1, MINT2, MINT3; and (4) activation of oncogenes such as RAS and BRAF.15
Pathogenesis
The colonic mucosa consists of epithelial cells arranged in cylindrical structures called crypts. The human colon contains about 10 millioncrypts. The cellular proliferation and migration in each of the crypts is believed to be tightly regulated, with the majority of cells arising from a small number of stem cells (around 4-6) at the bottom of the crypt, which migrate upward after division and are eventually shed into the lumen.16 Each crypt is renewed in 2 to 8 days, which makes colonic mucosa one of the organs with the most cell proliferation in the human body and, hence, a target for various genetic and epigenetic alterations as well as environmental mutagenesis.7
Traditionally, the majority of colorectal carcinomas were believed to evolve from adenomatous polyps, which transform into an advanced adenoma with high-grade
dysplasia and then progress to an invasive cancer often referred to as the adenoma-to-carcinoma sequence.15 However, 2 other pathways, alternative and serrated,
have been described, and CRCs are now regarded as complex malignancies with a wide array of genetic and epigenetic mutations.17
About 70% to 85% of CRCs generally develop from chromosomal instability resulting from inactivation of tumor suppressor genes (APC gene, p53, etc). About 15% of cases are attributed to the failure of the DNA mismatch repair system either by germline/somatic mutations or by epigenetic silencing of gene transcription by CpG island methylation.18 Mutation in the BRAF or K-ras oncogenes are also believed to promote carcinogenesis.15 All these changes are believed to give rise to a precursor microscopic mucosal lesion that precedes the development of macroscopic adenomas.17
Clinical Features
The clinical features of CRCs can be widely variable, from incidental findings during screening colonoscopy to intestinal obstruction. The most common clinical presentation is rectal bleeding, followed by weight loss, abdominal pain, constipation, or diarrhea.19 The likelihood of CRC is higher with the combination of rectal bleeding and weight loss as well as rectal bleeding and change in bowel habits. Other clinical features may include bloating, abdominal pain, or anemia.20 (See Table 1.)
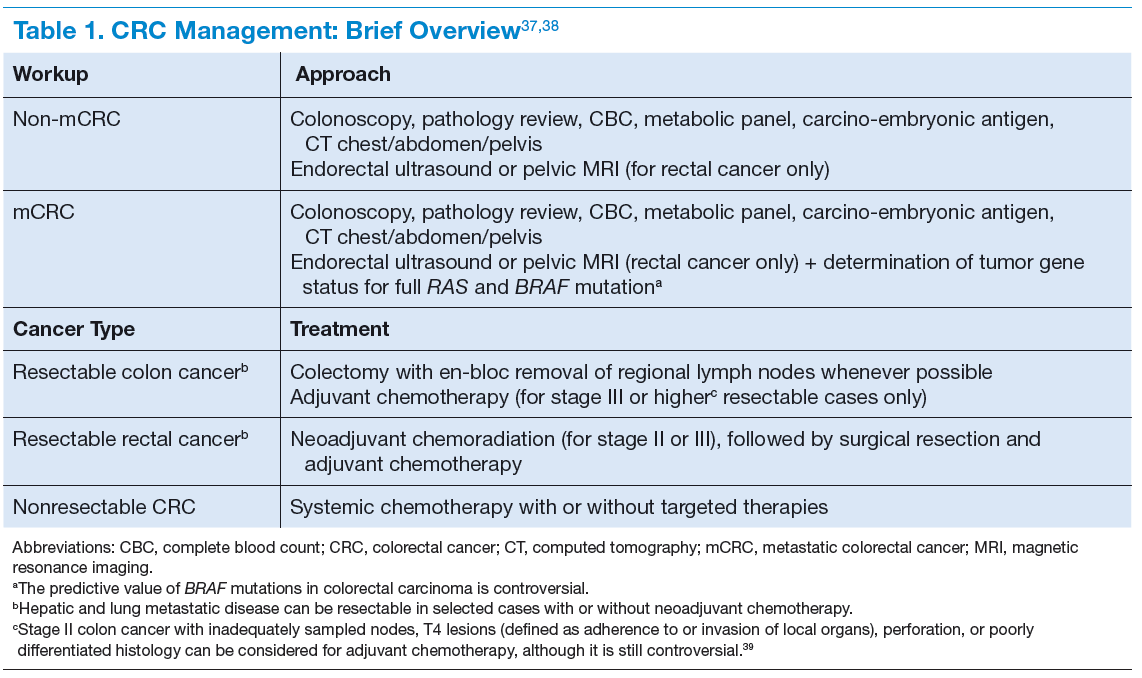
Targeted Therapy in CRC
The targeted therapies in CRC include (1) the antivascular endothelial growth factor-A (anti–VEGF-A) antibody bevacizumab; (2) the VEGF-A, VEGF-B, and placental growth factor inhibitor aflibercept; (3) the multikinase inhibitor regorafenib; and (4) the anti-epidermal growth factor receptor (anti-EGFR) antibodies cetuximab and panitumumab.
Bevacizumab is an anti-VEGF monoclonal antibody. Vascular endothelial growth factor promotes angiogenesisnecessary for tumor growth. Bevacizumab was approved by the FDA in February 2004 as a first-line treatment in combination with IFL (irinotecan plus 5-fluorouracil [5-FU]/leucovorin) regimen and in June 2006 as a second-line treatment in combination with 5-FU–based chemotherapy for patients with mCRC. In January 2013, bevacizumab was approved for use in combination with fluoropyrimidine–irinotecan- or fluoropyrimidine–oxaliplatin-based chemotherapy for the treatment of patients with mCRC whose disease has progressed while on first-line treatment with a bevacizumab-containing regimen.21 (See Table 2.)
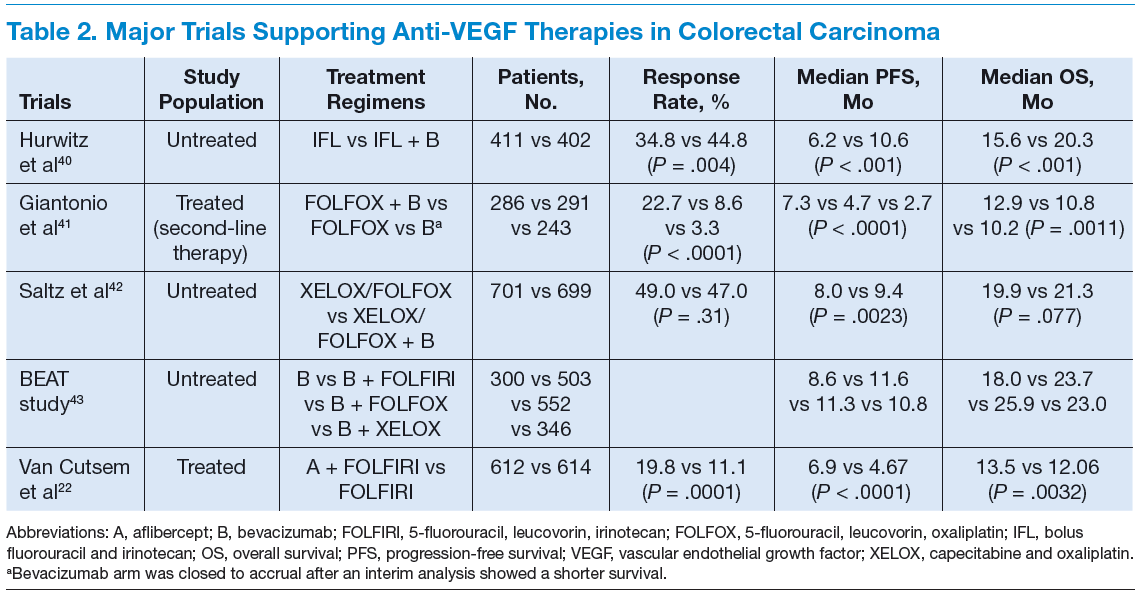
Aflibercept is a recombinant fusion protein, containing VEGF-binding portions from the extracellular domains of human VEGF receptors 1 and 2, fused to the Fc portion of human immunoglobulin IgG1 that blocks the activity of VEGF-A, VEGF-B, and placental growth factor by acting as a high-affinity ligand trap to prevent these ligands from binding to their endogenous receptors.22 It was approved by the FDA in August 2012 for use in combination with FOLFIRI (5-FU, leucovorin, irinotecan) for the treatment of patients with mCRC that is resistant to or has progressed following an oxaliplatin‑containing regimen.23
Cetuximab (a chimeric IgG1 anti-EGFR monoclonal antibody) and panitumumab (a human IgG2 anti-EGFR monoclonal antibody) have been shown to improve OS
and progression-free survival (PFS) in up to 20% of cases, either alone or in combination with chemotherapy.24,25 The FDA approved cetuximab in July 2012 for use in
combination with FOLFIRI for first-line treatment of patients with K-ras mutation-negative (wild-type), EGFRexpressing mCRC.26 The FDA approved panitumumab in
September 2006 for the treatment of patients with EGFRexpressing metastatic colorectal carcinoma with disease progression on or following FOLFOX (5-FU, leucovorin, oxaliplatin)/FOLFIRI.27 (See Table 3.)
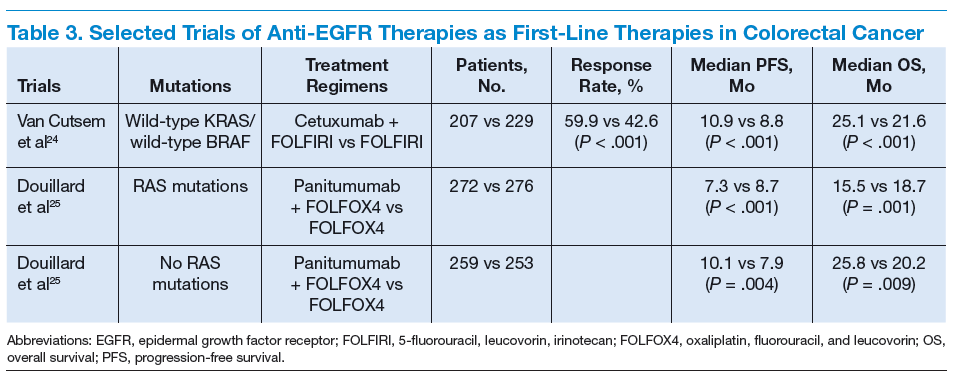
KRAS, a member of the rat sarcoma virus (ras) gene family of oncogenes, encodes for a small G protein downstream of EGFR. KRAS is mutated in CRC in up to 37% cases, resulting in activation of the different downstream signaling pathways.15,28 Therefore, KRAS mutations predict resistance to anti-EGFR therapy.28,29 Testing for KRAS mutation prior to treatment with cetuximab or panitumumab leads to targeted us of the very costly monoclonal antibodies and hence is considered a cost-effective practice.30
Even in cases with wild-type KRAS mutation, response to anti-EGFR therapies is seen in only less than half of patients.31 Up to 17% of tumors with wild-type for KRAS exon 2 at codons 12 and 13 can have a mutation in another of the ras pathway genes (eg, KRAS exon 3, 4 and NRAS exon 2, 3, 4).25 Mutations in the BRAF oncogene have been described in up to 13% of colorectal carcinoma cases.15 The data to suggest a lack of antitumor activity from anti-EGFR therapies in the presence of BRAF V600E mutation are still limited, but BRAF mutation is considered a poor prognostic factor.
A recent trial involving 1,137 patients with KRAS exon 2 wild-type mCRC randomly assigned to cetuximab or bevacizumab with standard chemotherapy (FOLFOX or FOLFIRI) found OS of 29.9 vs 29.0 months and median PFS 10.4 vs 10.8 months for cetuximab and bevacizumab, respectively.32 The OS for 5-FU–based therapies was about 11 months.33
Regorafenib is an oral multikinase inhibitor of angiogenic, stromal, and oncogenic receptor protein kinases, including those involved in the regulation of tumor angiogenesis (eg, VEGFR1-3 and TIE2 [tyrosine kinase with immunoglobulin and epidermal growth factor homology domain 2]), tumor microenvironment (plateletderived growth factor receptor-β and fibroblast growth factor receptor 1), as well as tumor oncogenesis (KIT, RET, RAF1, BRAF, BRAF V600E).34 A randomized phase 3 study involving 760 patients with documented progressive mCRC found a higher median OS with regorafenib vs placebo (6.4 mo vs 5.0 mo, P = .0052) and a
higher PFS (2.0 mo vs 1.7 mo, P < .0001).35 The FDA approved regorafenib in September 2012 for the treatment of patients with mCRC who have been previously treated with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, with an anti-VEGF therapy and, if KRAS wild-type, with an anti-EGFR therapy.36
Conclusion
Targeted therapies in conjunction with newer chemotherapies have improved outcomes in metastatic colorectal carcinoma compared with those of conventional therapy (29 mo vs 11 mo).
Author disclosures
Peter T. Silberstein, MD, reports receiving payment for lectures from Bristol Myers and Celgene in the past. Drs. Khanal and Upadhyay report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.