Ocular manifestations of small-vessel vasculitis





ABSTRACTOphthalmic manifestations of vasculitis can be orbital, ocular (affecting the globe), or intraocular. Orbital inflammation manifests as sudden onset of pain, erythema, and proptosis, and can be sight-threatening. In the globe, red eye is typical in both episcleritis and scleritis. Episcleritis is usually otherwise asymptomatic with blanching upon instillation of topical phenylephrine, whereas scleritis is painful and does not blanch. Infectious and rheumatic diseases are present in nearly 50% of patients with scleritis. The symptoms of keratitis are similar to those of scleritis; superficial keratitis is benign but peripheral ulcerative keratitis can be sight-threatening. Anterior uveitis is the most frequent ocular manifestation of Behçet disease. Approximately 30% of patients with granulomatosis with polyangiitis (Wegener’s granulomatosis) have ocular involvement, with orbital disease being most common. With ophthalmic manifestations of vasculitis, tissue biopsy of any site that is amenable to biopsy is recommended. Biopsy must be interpreted within the context of treatment.
We have long understood that vasculitic conditions have various clinical manifestations. The Chapel Hill Consensus Conference classification of systemic vasculitis in 19941 contributed significantly to our understanding of the spectrum of vasculitides and their manifestations, enhancing our diagnostic ability and the likelihood of appropriate treatment.
The ophthalmic manifestations of vasculitis are protean and nonspecific, and should be considered in the overall context of the disease. Patients should be evaluated with the following questions in mind:
- Are the manifestations related to the vasculitis itself?
- Are the manifestations a result or complication of therapy?
- Are the manifestations signs of a completely unrelated and superimposed condition?
This article reviews the three areas of ocular inflammation related to vasculitis and comments on the role of tissue biopsy in the management of these patients.
THREE AREAS OF OCULAR INFLAMMATION
Orbital inflammation
Orbital disease can affect the lacrimal gland (inflammatory dacryoadenitis), extraocular muscles (orbital myositis), and the orbital soft tissues (inflammatory orbital pseudotumor). Orbital inflammation is characterized by relatively sudden onset (within days) of pain, erythema, and proptosis. Diplopia and visual loss from either compression or inflammation of the optic nerve or nerve sheath may be present. Depending upon the structures involved and the degree of involvement, orbital inflammation can be sight-threatening.
Either computed tomography or magnetic resonance imaging should be performed to assess orbital or extraorbital involvement. The orbital structures are particularly amenable to biopsy, which, in this author’s opinion, should be performed whenever possible. The biopsy may need to be interpreted within the context of previous or concurrent immunosuppressive therapy, which can alter the histologic picture, minimize inflammation, and make detection of vasculitis difficult. In addition to identifying inflammation, biopsy helps to identify fungal infection or lymphoma that can follow prolonged immunosuppressive therapy.
Treatment of orbital inflammation requires corticosteroid therapy or some other type of systemic immunosuppression.
Ocular, or globe, inflammation
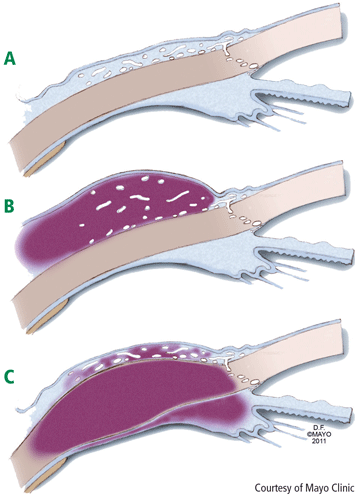
Episcleritis: observation or topical therapy. Episcleritis usually manifests as an otherwise asymptomatic red eye with typical sector-shaped inflammation. Pain is generally not an issue, although patients often report that the eye does not feel normal. Vision is unaffected and there is no potential threat to sight.
The slit-lamp examination shows dilated vessels in the episcleral tissues that blanch after instillation of a drop of 10% phenylephrine. Simple observation may be the best management course, but topical nonsteroidal anti-inflammatory drugs (NSAIDs) or topical corticosteroids may help some patients who have discomfort. There is probably a spectrum of disease in that some patients may have either severe episcleritis or mild scleritis (Figure 1B). At times it can be difficult to differentiate between severe episcleritis and mild scleritis. Although scleritis generally requires systemic therapy, topical therapy is justified for mild scleritis. Episcleritis is associated with systemic disease in approximately 36% of patients.2–4
Scleritis: may be sight-threatening; requires systemic therapy. Scleritis characteristically presents with intense pain and a red eye.3,5–7 Patients may be sensitive to light and their vision may be compromised. Cataracts and glaucoma can complicate the course of scleritis.
With slit-lamp examination, the redness does not blanch upon instillation of topical 10% phenylephrine as it does with episcleritis. The adjacent cornea may also be affected (Figure 1C). Healed scleritis leaves an area of thinned sclera that appears as a visible blue spot, so if the patient’s history includes red eye with pain and a blue area is visible, the clinician can be confident that a prior episode of scleritis occurred.
Scleritis can be anterior or posterior, and the implications are slightly different for each type. Anterior scleritis can be subclassified as diffuse, nodular, or necrotizing. The necrotizing type can be characterized by painful inflammation or, in the case of scleromalacia perforans, no inflammation and no pain. Posterior scleritis may have minimal pain.
Akpek et al5 reported on a group of 243 patients with scleritis (average age, 52 years; range, 5 to 93 years) who were followed for an average of 1.7 years (range, 0 to 16.6 years). An associated medical condition was present in 107 (44%) patients. Rheumatologic conditions accounted for 37%, with rheumatoid arthritis being most common; infectious disease, with herpes zoster ophthalmicus being most common, accounted for 7%. Of those with an associated medical condition, 78% had been diagnosed previously; the remaining 22% were diagnosed at presentation or the condition developed during follow-up.
Treatment typically requires systemic therapy with NSAIDs, but more often oral or intravenous corticosteroids or even methotrexate, mycophenolate mofetil, cyclophosphamide, or rituximab may be required. Patients with antineutrophil cytoplasmic antibody (ANCA)–positive disease may require more intensive therapy than those with ANCA-negative disease.
Keratitis: may be sight-threatening. Patients with keratitis should be evaluated in the same spirit as patients with scleritis (Figure 1C). Although many patients may have superficial keratitis, which is often related to a dry eye and has no prognostic significance, deep or peripheral ulcerative keratitis is not only consistent with systemic vasculitis but also sight-threatening. Symptoms similar to those observed with scleritis typically include severe pain and photophobia and, as with scleritis, treatment usually involves systemic therapy.