Use of chemodenervation in dystonic conditions





ABSTRACTDystonia, an uncommon movement disorder that causes sustained muscle contractions and painful body positions, is a difficult diagnostic challenge; misdiagnosis is common. Classification may include etiology, area of physical involvement, or age of onset. Bodily distribution is varied, and dystonias can present as primary (genetic) or secondary (caused by other disease processes or use of neuroleptic drugs). Although there is no cure, the use of botulinum toxins for chemodenervation provides symptomatic relief and is considered the treatment of choice in focal dystonia. The dose of botulinum toxin may be titrated to provide significant relief for 12 weeks or more.
Dystonia is a movement disorder in which involuntary sustained muscle contractions cause twisting movements that place the body in abnormal, sometimes painful, positions. Dystonia is believed to arise from an abnormality in the basal ganglia and an inherent or acquired defect in the processing of neurotransmitters.1
Dystonia is uncommon, although its exact prevalence is unknown. Nutt et al concluded that at least 250,000 people were affected by idiopathic dystonia in the United States, but prevalence is likely higher because misdiagnosis is not uncommon.2 A more recent European study found the prevalence of primary dystonia in the general population aged 50 years or more to be 732 per 100,000.3 The Epidemiological Study of Dystonia in Europe (ESDE) Collaborative Group found that the estimated prevalence of cervical dystonia was 50 to 200 per 1 million individuals.4 Also known as spasmodic torticollis, this is the most commonly diagnosed form of focal dystonia.
CLASSIFICATION OF DYSTONIA
Accurate classification of dystonia is important, since this informs approaches to management as well as prognosis. The three most important means by which dystonia is classified are (1) etiology, including primary dystonia, which encompasses a variety of genetic variables, and secondary dystonia; (2) bodily distribution of symptoms; and (3) age at onset.
Etiology
Most primary or idiopathic dystonia appears to be hereditary. Early-onset primary dystonia is most frequently caused by a mutation in the DYT1 gene, although other genetic mutations are possible.5 Patients with primary dystonia have no other underlying disorder; involuntary muscle contractions are the sole symptom. A thorough history should include a review of perinatal and early developmental history, prior neurologic illness, and exposure to drugs known to cause acquired dystonia. Physical examinations (encompassing intellectual, pyramidal, cerebellar, and sensory domains) and laboratory tests reveal no specific cause for the dystonic symptoms. Primary dystonia is also most frequently action-induced; at rest, the affected body region may appear to be normal.
Secondary dystonia occurs as a symptom of another disease process. Multiple sclerosis or any one of several hereditary neurologic disorders, such as Wilson disease, may be implicated. Secondary dystonia also may result from trauma to the brain, as might occur during an automobile accident; from heavy-metal or carbon monoxide poisoning; or as an adverse effect of medication. It may be psychogenic or related to Parkinson disease or Parkinson-plus syndromes, a group of neurodegenerative disorders with parkinsonian features. Tardive dystonia, the most common adult form of secondary dystonia, may occur follow ing exposure to certain neuroleptic drugs; tardive dystonia is a type of tardive dyskinesia that describes any involuntary neurologic movement disorder.
Bodily distribution
Dystonia is further classified by location of symptoms. Focal dystonias, which are usually primary dystonias, describe symptoms that are limited to a region of the body, such as a specific arm. There are several variations. Cervical dystonia affects the head and neck, is the most common adult-onset dystonia, and affects more women than men. Blepharospasm, or involuntary contractions of the eyelids, potentially leads to extended eye closure and functional blindness and often involves other facial muscles. Laryngeal dystonia affects the muscles in the larynx. Limb dystonia, such as writer’s or musician’s cramp, affects muscles in the arm, hand, leg, or foot. Limb dystonia is often task-specific action dystonia, and can be primary or secondary.
Segmental dystonia describes a group of involved muscles that are contiguous, such as cranial to neck to cervical to arm. Oromandibular dystonia, affecting the face, mouth, and jaw, often with unusual tongue movements (ie, lingual dystonia), is a type of segmental dystonia, although some consider it a focal dystonia. Meige syndrome is the combination of blepharospasm and oromandibular dystonia. Certain limb and cranial dystonias are considered segmental dystonias. Dystonia that affects two or more noncontiguous muscle groups in different parts of the body is multifocal. Hemi dystonia describes unilateral symptoms.
Symptoms that have advanced from a focal presentation to affect additional regions of the body characterize generalized dystonia. The symptoms potentially advance to include the trunk and limbs. The muscular contractions are usually sustained, are often both repetitive and painful, and worsen with activity.6 In severe cases, muscular contractions may occur even while resting. Early-onset myoclonus dystonia is a generalized hereditary dystonia whose symptoms include dystonic contractions of the neck and shoulders and rapid jerking movements.7 Of note diagnostically, early-onset dystonia in a leg typically begins at age 8 to 9 years and is more likely than other early-onset presentations to progress to generalized dystonia. Early-onset dystonia that begins in an arm typically presents later, at age 12 to 14 years, and is less likely to progress to generalized dystonia. Late-onset dystonia (> 27 years of age), by contrast, rarely begins in a leg and tends to remain either focal or segmental.8
Age of onset
A third useful classification scheme identifies early-onset (childhood to young adult) and late-onset varieties of dystonia.
THE DIAGNOSTIC CHALLENGE
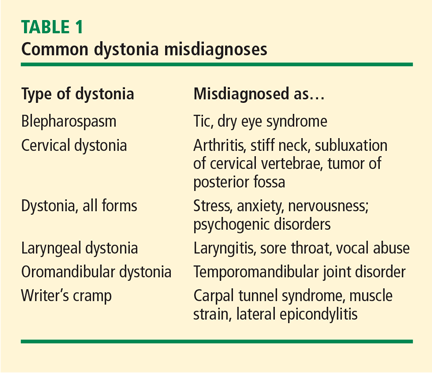
Consider primary dystonia if perinatal and developmental histories, intellect, strength, and perception of sensations are normal. There should be no prior history of neurologic illness or exposure to neuroleptic drugs whose adverse effects include secondary dystonia. In primary dystonia, diagnostic studies are negative and dystonia is the only symptom. If onset of symptoms is associated with activity, then primary dystonia should be considered. In the case of early- or late-onset limb dystonia, testing should be performed for the DYT1 gene. If the results are negative, then a trial for dopa-responsive dystonia should be undertaken with levodopa.
Consider secondary dystonia if the patient has been exposed to neuroleptic drugs, symptoms are distributed unilaterally, or the presentation is unusual for age or distribution of symptoms. For example, cranial dystonia in a child would raise the index of suspicion for secondary dystonia. If tardive dystonia is part of the differential diagnosis, consider magnetic resonance imaging (MRI), serum ceruloplasmin measurement, or slit-lamp diagnostic testing. Suspicion of a structural lesion affecting the central nervous system warrants examination with MRI, computed tomography, or angiography. Certain metabolic and neurologic hereditary disorders cause secondary dystonia, in which case dopa-responsive dystonia should be ruled out. Psychometric testing should also be considered.