
Rucaparib – second PARP inhibitor hits the market for ovarian cancer





What’s new, what’s important
The PARP inhibitor rucaparib’s approval for the treatment of BRCA1/2 mutant advanced ovarian was based on findings from 2 trials (Study 10 and ARIEL2) in which rucaparib showed complete or partial tumor shrinkage in more than half of the participants. It was approved with a companion diagnostic.
Patients in both studies had BRCA1/2 mutation-positive ovarian cancer and had progressed on 2 or more previous chemotherapy regimens. ARIEL2 also included patients with platinum-sensitive, resistant, and refractory disease. In Study 10, the ORR was 60%, and CR and PR were 10% and 50%, respectively, over a median DoR of 7.9 months; in ARIEL2, the ORR was 50%, and CR and PR were 8% and 42%, over a median DoR of 11.6 months (pooled analysis, median DoR of 9.2 months: ORR, 54%; CR, 9%; PR, 45%).
Across studies, the most common adverse events included nausea, fatigue, vomiting, and anemia, and the most common grade 3/4 AEs were anemia, fatigue/asthenia, and increased alanine aminotransferase or aspartate aminotransferase levels, with 8% of patients discontinuing therapy because of AEs. The recommended dose is 600 mg PO twice daily. Prescribing physicians should be aware of the risk for MDS or acute myeloid leukemia, and for embryofetal toxicity. Complete blood count should be monitored at baseline then monthly. If hematologic toxicities occur, treatment should be interrupted and blood counts monitored. Pregnant women and those of reproductive age should be advised about the risk to the fetus or the need for effective contraception during treatment and 6 months after the last dose.
Correspondence Jame Abraham, MD, FACP (abrahaj5@ccf.org)
Citation JCSO 2017;15(2):62-64
©2017 Frontline Medical Communications
doi https://doi.org/10.12788/jcso.0333
Related articles
Slow and steady progress in managing gynecologic cancer
Hereditary breast and ovarian cancer: risk assessment in minority women and provider knowledge gaps
Submit a paper here
Rucaparib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with BRCA1/2 mutant advanced ovarian cancer in January this year, making it the second drug in its class for this indication. It is a poly(ADP-ribose) polymerase inhibitor that works by blocking the repair of damaged DNA in cancer cells and triggering cell death.
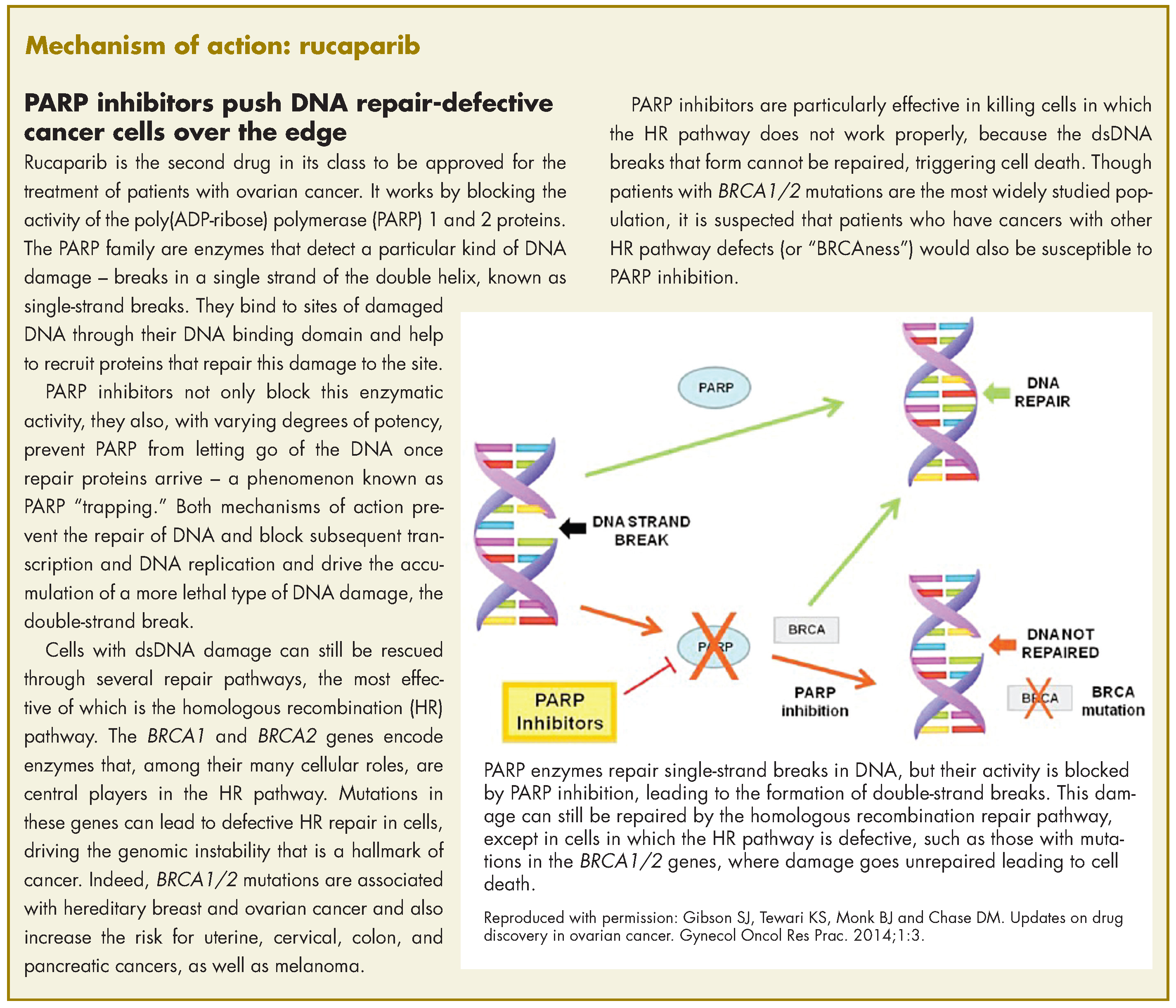
The approval was based on findings from 2 single-arm clinical trials in which rucaparib led to complete or partial tumor shrinkage in more than half of the patients enrolled. A pooled analysis included 106 patients from the phase 2 trials, Study 10 (NCT01482715; N = 42) and ARIEL2 (NCT01891344; N = 64), in which patients with BRCA1/2 mutation-positive ovarian cancer who had progressed on 2 or more previous chemotherapy regimens, received 600 mg rucaparib twice daily.
Study 10 included only patients with platinum-sensitive disease and eligible patients were aged 18 years or older, with a known deleterious BRCA mutation, evidence of measurable disease as defined by Response Evaluation Criteria in Solid Tumors (version 1.1), sufficient archival tumor tissue, histologically confirmed high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer and relapsed disease confirmed by radiologic assessment. Meanwhile, ARIEL2 had similar eligibility criteria, except that patients with platinum-sensitive, resistant, and refractory disease were included.
,Both studies excluded patients with active second malignancies, and for those with a history of prior cancer that had been curatively treated, no evidence of current disease was required and chemotherapy should have been completed more than 6 months or bone marrow transplant more than 2 years before the first dose of rucaparib. Patients who had previously been treated with a PARP inhibitor, with symptomatic and/or untreated central nervous system metastases, or who had been hospitalized for bowel obstruction within the previous 3 months, were also ineligible.
Across the 2 trials, the median age of trial participants was 59 years, 78% were white, and all had an Eastern Cooperative Oncology Group performance status of 0 (fully active, able to carry on all pre-disease performance without restriction) or 1 (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature). Both trials used a surrogate endpoint for approval, measuring the percentage of patients who experienced complete or partial tumor shrinkage, the overall response rate (ORR), while taking rucaparib.
In Study 10, the ORR was 60%, including a complete response (CR) rate of 10% and a partial response (PR) rate of 50%, over a median duration of response (DoR) of 7.9 months, while in ARIEL2, the ORR was 50%, including a CR of 8% and a PR of 42%, over a median DoR of 11.6 months. The pooled analysis demonstrated an ORR of 54%, CR of 9% and PR of 45%, over a median DoR of 9.2 months. In separate data reported in the prescribing information, the ORR as assessed by independent radiology review was 42%, with a median DoR of 6.7 months, while ORR according to investigator assessment was 66%. In all analyses, the response rate was similar for patients having BRCA1 versus BRCA2 gene mutations.
Safety analyses were performed in 377 patients across the 2 studies who received 600 mg rucaparib twice daily. The most common adverse events (AEs) of any grade included nausea, fatigue, vomiting, anemia, abdominal pain, dysgeusia, constipation, decreased appetite, diarrhea, thrombocytopenia, and dyspnea. The most common serious AEs (grade 3 or 4) were anemia (25%), fatigue/asthenia (11%), and increased alanine aminotransferase or aspartate aminotransferase levels (11%). Overall, 8% of patients discontinued treatment because of AEs.
The recommended dose according to the prescribing information is 600 mg, in the form of two 300-mg tablets taken orally twice daily with or without food. Phy
If hematologic toxicities occur while taking rucaparib, treatment should be interrupted and blood counts monitored until recovery and failure to recover to grade 1 or higher after 4 weeks should prompt referral to a hematologist for further investigation, while confirmed diagnosis of myelodysplastic syndromes or acute myeloid leukemia should lead to discontinuation of rucaparib. Pregnant women and those of reproductive potential should be advised of the potential risk to a fetus or the need for effective contraception during treatment and for 6 months after the last dose of rucaparib.
Rucaparib is indicated only for the treatment of patients with confirmed BRCA1/2 mutations, so the drug was approved in conjunction with a companion diagnostic. FoundationFocus CDxBRCA is the first next-generation sequencing-based test to receive FDA approval and detects the presence of deleterious BRCA gene mutations in tumor tissue samples. Rucaparib is marketed as Rubraca by Clovis Oncology Inc, and the companion diagnostic by Foundation Medicine Inc.
